TO THE EDITOR:
Delayed platelet recovery and secondary thrombocytopenia, defined as a secondary decrease in platelet count not due to disease relapse after initial platelet recovery, occur in 5% to 25% of patients after hematopoietic stem cell transplantation (HSCT) and are of adverse prognostic significance.1-3 The usual causes are insufficient stem cell dose, infections, graft-versus-host disease (GvHD), and drug toxicity. Platelet transfusion remains a mainstay of therapy, whereas the role of thrombopoietin (TPO) receptor agonists is poorly known in this setting.4 Romiplostim (Nplate; Amgen, Thousand Oaks, CA) is an Fc-peptide fusion protein (peptibody) that leads to increased platelet production via the TPO receptor. Romiplostim is injected subcutaneously on a weekly basis, which is convenient in the context of HSCT, especially in patients with gut GvHD. In this phase 1/2 multicenter single-arm open trial, we investigated the safety and efficacy of romiplostim for patients with transfusion-dependent thrombocytopenia after allogeneic HSCT (NCT01980030).
Adult patients undergoing standard-of-care allogeneic HSCT were eligible ≥45 days after transplantation if they had a platelet count ≤20 × 109/L that was sustained for 7 days (≤50 × 109/L with a history of bleeding) or if they were platelet transfusion dependent. Patients were excluded if they had abnormal liver function tests, serum creatinine ≥176.8 μmol/L, had prior venous thrombosis, or were transplanted for myelodysplastic syndrome (theoretical concern at the time of the study that TPO receptor agonists may stimulate the progression of myelodysplastic syndrome). Patients were given weekly romiplostim for 12 weeks, with intrapatient weekly dose escalation from 1 µg/kg to a maximum dose of 10 µg/kg. The primary safety end point was the incidence of any grade 3 or 4 adverse events (AEs) after HSCT, excluding those expected related to HSCT, as well as clinically significant bleeding events. The primary efficacy end point was the time to reach a platelet count >50 × 109/L without a platelet transfusion. Secondary end points were the durable platelet response, defined as a platelet count ≥50 × 109/L for 8 consecutive weeks independent of platelet transfusions after transplant, as well as the 1-year cumulative incidence (CumI) after HSCT of GvHD, relapse, and nonrelapse mortality rates. The CumI of absolute neutrophil counts >1000 × 109/L and the CumI of hemoglobin ≥10 g/dL were also analyzed during the study period. All patients had a bone marrow biopsy before treatment and at 1 year. The protocol was approved by the Saint Louis Hospital Institutional Review Board and by French regulators. All patients provided written informed consent, in accordance with the Declaration of Helsinki. The statistical method was based on an intent-to-treat analysis. CumIs were estimated using nonparametric methods; deaths prior to the event of interest defined competing risks. Point estimates are reported with 95% confidence intervals (95% CIs).
Standard treatment in case of delayed platelet recovery and secondary thrombocytopenia is platelet transfusion. Literature on the use of TPO receptor agonists in this setting is sparse and mainly limited to single case reports or retrospective case series.3,5-9 Here, we report a prospective clinical trial of 24 patients from 4 French HSCT hospitals (Necker, Saint Antoine, Saint Louis, and Pitié-Salpétrière hospitals) belonging to Assistance Publique Hopitaux de Paris, who were treated between April of 2013 and November of 2015. Patient characteristics, disease characteristics, and main transplantation outcomes under romiplostim are summarized in Table 1. Ten patients had delayed engraftment at the time of romiplostim initiation, whereas 14 exhibited secondary thrombocytopenia due to GvHD and/or concomitant infections. The median time between HSCT and romiplostim initiation was 85 days (interquartile range [IQR], 63-117; range, 42-259). Nineteen patients received all 12 investigational injections of romiplostim, whereas 5 patients dropped out of the study: 3 because of death (1 from a posttransplantation Epstein-Barr virus–related lymphoproliferative disorder, 1 who relapsed, and 1 from septic shock), 1 because of a boost of donor stem cells, and 1 with a platelet count >50 × 109/L after 8 injections. Six patients experienced a total of 21 AEs (grade 3, n = 10; grade 4, n = 11; according to Common Terminology Criteria for Adverse Events). Hematological complications appeared in 4 patients (2 neutropenia, 1 anemia, and 1 pancytopenia). Liver dysfunction was present in 2 patients (supplemental Table 1, available on the Blood Web site). The 6-month CumI of AEs was 25.2% (95% CI, 7.3-43.2). Overall, romiplostim was considered well tolerated (the association of the reported AEs with the drug was considered to be unlikely related). However, 5 patients dropped out of the study, including 3 patients who died, but only 6 patients experienced grade 3 or 4 AEs (supplemental Table 1). Thus, we cannot exclude that AEs were underreported. Importantly, bone marrow biopsies performed before romiplostim administration, as well as at 12 weeks and 1 year after treatment initiation, did not show any increase in marrow fibrosis, which was reported with the use of TPO agents.10
With regard to transplantation outcomes (Table 1), a total of 17 and 12 patients experienced acute GvHD and chronic GvHD, respectively, with 100-day CumI of acute GvHD of 67% (95% CI, 47-87) and 1-year CumI of chronic GvHD of 55% (95% CI, 32-78). No bleeding events or thrombotic complications were recorded, and the 1-year CumI of relapse was 13% (95% CI, 0-27), as expected in such a cohort of HSCT patients. Six patients died during the study (1 relapsed, 1 from posttransplantation Epstein-Barr–related lymphoproliferative disorder, 1 from lung aspergillosis, 1 from septic shock, and 2 from acute respiratory distress syndromes of unknown origin). Nonrelapse mortality was 21% (95% CI, 7-42), as expected after HSCT, in our study population (>2/3 myeloablative conditioning regimen). Overall, romiplostim was very well tolerated in the setting of thrombocytopenia post-HSCT, and we did not observe any unexpected toxicity.
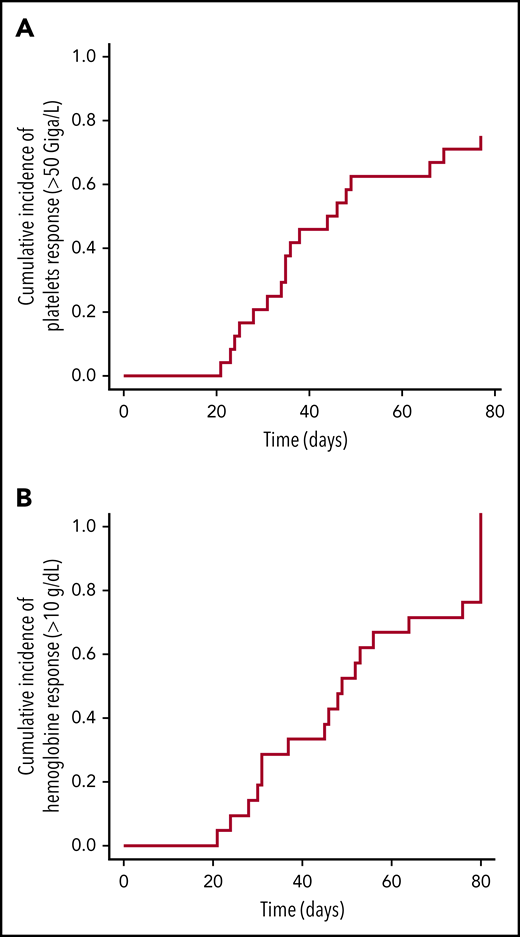
With regard to efficacy, 18 patients with thrombocytopenia responded to treatment. The median time to reach a platelet count >50 × 109/L free of platelet transfusion was 45 days (IQR, 29-41; range, 21-77) (Figure 1A), with required doses of 5 μg/kg (IQR, 4-6.8; range, 1-11). A hemoglobin response was also observed in 21 of 22 patients who had levels <10 g/dL before the first injection (Figure 1B), and neutrophil counts improved in the 4 patients who had <1000 × 109/L before treatment initiation, which suggest an effect similar to the use of TPO receptor agonists (ie, eltrombopag) in patients suffering from refractory aplastic anemia.11,12 Sixteen patients achieved a durable platelet response (8 consecutive weeks, independent of platelet transfusion). All patients showed donor chimerism at the end of the study (with the exception of those who relapsed). However, because of the limited number of cases, we were not able to identify factors predictive of a romiplostim response. In the absence of a control arm, we cannot exclude that platelet recovery was due to spontaneous improvement. Nevertheless, the response rate is very encouraging, and the time to respond is ∼45 days. In a study using CD34-selected cells to treat patients in a similar hematological situation, the median time to trilineage recovery was 3 months.13 Moreover, access to romiplostim is easy.
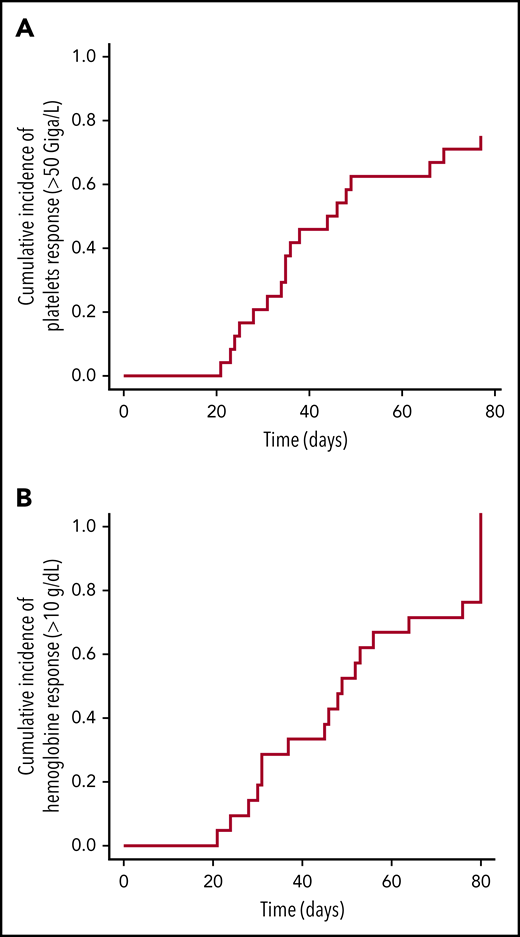
Longitudinal platelet and hemoglobin improvements in patients who received romiplostim after HSCT. (A) CumIs during the study period of platelets counts ≥50 × 109/L (all patients). (B) CumIs of hemoglobin ≥10 g/dL (22 patients at time of first injection). Starting point for analysis is time of study enrollment.
Longitudinal platelet and hemoglobin improvements in patients who received romiplostim after HSCT. (A) CumIs during the study period of platelets counts ≥50 × 109/L (all patients). (B) CumIs of hemoglobin ≥10 g/dL (22 patients at time of first injection). Starting point for analysis is time of study enrollment.
In conclusion, romiplostim can be safely administered to patients with transfusion-dependent thrombocytopenia after allogeneic HSCT, and it was also able to improve platelet count using a dose ≥5 μg/kg to reach platelet counts >50 × 109/L at a median of 45 days. Therefore, romiplostim is particularly interesting considering the increasing use of alternative-donor HSCT and the high rate of toxicity related to delayed platelet recovery and secondary thrombocytopenia after HSCT; this needs confirmation through a randomized prospective phase 3 clinical trial.
Presented in abstract form at the 58th annual meeting of the American Society of Hematology, San Diego, CA, 3 December 2016.
Readers have access to the protocol by sending an email to the corresponding author.
The online version of this article contains a data supplement.
Acknowledgments
The authors thank colleagues who actively participated in the study, and they would particularly like to thank the patients and their families for kindly agreeing to take part in this research.
This work was supported in part by Amgen.
Authorship
Contribution: R.P.d.L., S.C., and G.S. conceived and designed the study, collected, assembled, analyzed, and interpreted data, and wrote the manuscript; R.P.d.L., A.L.R., F.S., L.S., D.M., F.S.d.F., T.C., N.D., M.T.R., S.N., M.M., and G.S. provided study materials and patients; and R.P.d.L., S.C., A.L.R., F.S., L.S., D.M., F.S.d.F., T.C., N.D., M.T.R., S.N., M.M., and G.S. approved the final version of the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Régis Peffault de Latour, Service d’Hématologie Greffe, Hôpital Saint Louis, Université de Paris, Paris, France; e-mail: regis.peffaultdelatour@aphp.fr.