Abstract
Traditionally, primary immune deficiencies have been defined based on increased susceptibility to recurrent and/or severe infections. However, immune dysregulation, manifesting with autoimmunity or hyperinflammatory disease, has emerged as a common feature. This is especially true in patients affected by combined immune deficiency (CID), a group of disorders caused by genetic defects that impair, but do not completely abolish, T-cell function. Hypomorphic mutations in the recombination activating genes RAG1 and RAG2 represent the prototype of the broad spectrum of clinical and immunological phenotypes associated with CID. The study of patients with RAG deficiency and with other forms of CID has revealed distinct abnormalities in central and peripheral T- and B-cell tolerance as the key mechanisms involved in immune dysregulation. Understanding the pathophysiology of autoimmunity and hyperinflammation in these disorders may also permit more targeted therapeutic interventions.
Introduction
Historically, primary immune deficiencies (PIDs) have been defined based on an increased susceptibility to infections. However, it has become increasingly evident that immune dysregulation is an important component of several forms of PID and, in fact, represents the main manifestation in “Diseases of immune dysregulation” listed in the most recent classification of PIDs from the International Union of Immunological Societies (IUIS).1 In addition to these disorders, other forms of PIDs may include autoimmunity or excessive inflammation as part of the clinical phenotype. In a survey of 2183 patients reported to the French PID registry, 571 (26.2%) had ≥1 autoimmune or inflammatory condition, with common variable immune deficiency and combined immune deficiencies (CIDs) being associated with the highest risk of immune dysregulation2 .
According to the IUIS classification, defects resulting in combined defects of cellular and humoral immunity include a heterogeneous group of 49 disorders characterized by a reduced number and/or function of T cells.1 Based on the severity of the T-cell defect, inborn errors of cellular and humoral immunity are further classified into “severe combined immune deficiencies” (SCIDs) and other CIDs, in which the immunological defect is generally less profound.1 The Primary Immune Deficiency Treatment Consortium has developed diagnostic criteria for typical and atypical forms of SCID, with the latter having slightly higher T-cell count and function.3 Diagnostic criteria for CIDs other than SCID have been proposed by the European Society for Immune Deficiencies.4 A different distribution of genotypes has been reported in SCID and CID1,5,6 ; however, mutations in the same gene may associate with a broad spectrum of clinical and immunological phenotypes, as best exemplified by defects in the recombinase activating genes RAG1 and RAG2.7 Immune dysregulation is more commonly observed in patients with CID than in those with SCID2 ; in the latter, it is more frequent among patients with atypical SCID (AS),8 indicating that subnormal (but not absent) T-cell mediated immunity is a permissive factor. Manifestations of immune dysregulation in CID may include systemic and organ-specific autoimmunity and inflammatory complications; however, autoimmune cytopenias are more frequently observed.2,6,7 Importantly, immune dysregulation represents a negative prognostic factor for survival in patients with CID, even for patients who are eventually treated by hematopoietic stem cell transplantation (HSCT).2 Altogether, these data indicate the importance of prompt recognition and treatment of immune dysregulation in patients with CID. Finally, the study of these patients is illustrative of how different degrees of impairment of T- and B-cell development and function translates in defective adaptive immune responses against nonself antigens, as well as perturbs immune homeostasis by affecting central and peripheral mechanisms of immune tolerance.9 Here, we will review the clinical manifestations, the laboratory findings, and the pathophysiology of immune dysregulation associated with RAG deficiency and other forms of CID (Table 1).
Immune dysregulation in RAG deficiency
The recombination activating proteins (RAG1 and RAG2) initiate the process of V(D)J recombination, allowing for the formation of a diverse repertoire of T-cell receptors (TCRs) in T cells and immunoglobulins (Igs) in B cells.10 This process is essential for the generation of polyclonal T and B cells with a broad antigen-recognition capacity.11,12 Biallelic RAG mutations were initially associated with SCID with absent T and B cells (T–B–SCID).13 Subsequently, the clinical presentation of RAG deficiency expanded to include Omenn syndrome (OS), in which residual RAG protein activity allows for the generation of oligoclonal T cells.14,15
Patients with OS present in infancy with life-threatening infections, erythroderma, lymphadenopathy, hepatosplenomegaly, eosinophilia, and severe hypogammaglobulinemia with increased IgE levels. They typically have absent B cells and expanded activated populations of autologous autoreactive T cells that infiltrate the skin, gut, liver, and other organs. Interestingly, within the same patient with OS, the T cells that infiltrate distinct organs carry distinct TCR specificities, suggesting tissue-specific self-antigen–driven expansion of T-cell clonotypes.16
Hypomorphic RAG mutations may also cause AS,17 in which patients have variable, but reduced, numbers of T and B cells with a decreased proportion of naive T cells. Patients with AS are highly susceptible to severe and opportunistic infections8 ; however, in contrast to typical SCID, they are at high risk for autoimmune manifestations, especially cytopenias.7,8 A distinct form of AS associated with oligoclonal expansion of TCRγδ+ T cells and an increased risk for autoimmune hemolytic anemia upon cytomegalovirus (CMV) infection has been reported in RAG deficiency.18,19
More recently, RAG mutations have also been detected in patients with delayed-onset disease exhibiting granulomas and/or autoimmune manifestations (CID-G/AI phenotype).7,8,20-25 In these patients, T and B cells are usually present, in vitro T-cell proliferation to mitogens is often normal or slightly reduced, and IgG levels and antibody responses to immunization may be variably preserved. In this group of patients, early-onset severe infections are infrequent, but late-onset immune dysregulation and infections cause significant morbidity.7,8 In particular, recurrent respiratory tract infections leading to bronchiectasis and chronic and recurrent viral infections due to herpesviruses and human papillomavirus are common in CID-G/AI.7,8
Overall, these observations show that immune dysregulation is a prominent manifestation of impaired RAG function that is especially common among patients exhibiting SCID or CID-G/AI.
In a cohort of 424 patients with RAG deficiency, autoimmunity and/or granulomatous lesions occurred in 6.5% of patients with OS, 45% of patients with AS, and 89% of patients with CID-G/AI and other delayed-onset phenotypes.7 Cytopenias were the leading autoimmune manifestation, with 55% (74/134) of AS and CID-G/AI patients presenting with autoimmune hemolytic anemia, immune thrombocytopenic purpura, or autoimmune neutropenia and 26% suffering from ≥2 autoimmune cytopenias. Other autoimmune phenomena occurred in skin, vasculature, gut, endocrine organs, and neuronal and kidney tissues. Along with these manifestations of autoimmunity, a broad range of autoantibodies, including neutralizing antibodies to interleukin-12 (IL-12), interferon-α (IFN-α), and IFN-ω, were detected in patients with CID-G/AI.26 Severe varicella was reported in some of these patients, suggesting that the presence of these anti-cytokine antibodies may increase the risk of infections or that, alternatively, varicella may trigger autoimmunity.
Granulomas, found in 35% of the patients with CID-G/AI, were predominantly localized in the skin, but were also observed in lungs, liver, soft tissues, spleen, gut, testis, bone marrow, and pancreas.7
In another series of 63 patients with RAG deficiency with prominent autoimmune and/or hyperinflammatory manifestations (most of whom belonged to the AS and CID-G/AI phenotypes), 84.1% had autoimmune cytopenias, 23.8% had granulomas, and 19% had skin manifestations.8 In this cohort, autoimmune cytopenias were often the presenting sign of disease and were frequently refractory to treatment with IV Ig, steroids, and rituximab. Similarly, granulomas, vasculitis, and enteropathy often failed to respond to first- and second-line immunosuppressive treatment, including steroids, IV Ig, cyclophosphamide, infliximab, cyclosporine, and sirolimus. Treatment-refractory autoimmunity and/or hyperinflammation were an indication of HSCT in 20 patients.
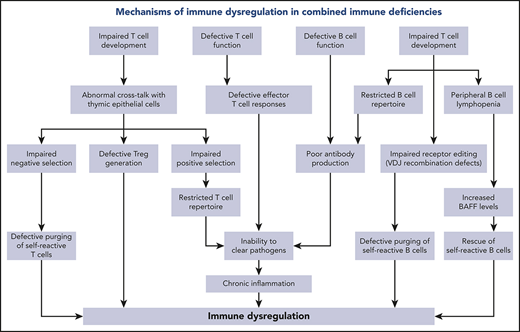
Various mechanisms contribute to the complexity and severity of immune dysregulation in patients with hypomorphic RAG mutations. In the normal thymus, developing T cells expressing self-reactive specificities are subjected to negative selection upon high-affinity interaction with thymic epithelial cells (TECs) and thymic dendritic cells presenting self-antigens in the context of major histocompatibility molecules. The first such wave of negative selection occurs in the cortex; depletion of T-cell clonotypes containing cysteine residues at the center of the TCR-CDR3 has recently been identified as a biomarker of this first wave of negative selection.27 A second wave of negative selection occurs in the thymic medulla, when autoreactive T cells binding with high-affinity self-peptides are deleted or, alternatively, are diverted to become regulatory T cells (Tregs). Importantly, developing T cells provide instructional signals to medullary TECs (mTECs), allowing their maturation and promoting expression of autoimmune regulator, a transcriptional activator that induces expression of tissue-restricted antigens by mTECs.28 A biomarker of this second wave of negative selection is represented by purging of TCR clonotypes containing hydrophobic amino acids at positions 6 and 7 of the TCR-CDR3.29 In contrast to what is observed in healthy individuals, the thymus of RAG-deficient patients has an abnormal architecture.23,30 Defective cross talk between TECs and the few thymocytes that are able to overcome the differentiation block results in impairment of negative selection. The high frequency of peripheral TCR clonotypes containing central cysteine27 is indicative of impairment of wave 1 deletion of self-reactive thymocytes in the thymus cortex. In the medulla, defective thymopoiesis causes impaired maturation of mTECs, as well as reduced expression of autoimmune regulator and tissue-restricted antigens.31 Peripheral T cells from RAG-mutated patients express TCR clonotypes with an increased frequency of hydrophobic amino acids at positions 6 and 7 of the CDR3, indicating impairment of the second wave of negative selection, as well.27,32 Tregs are present in low number, display a restricted repertoire,32,33 and have reduced suppressive function.33-35
Finally, the few oligoclonal T cells that egress the thymus undergo homeostatic proliferation in the periphery. In this setting, increased cytokine production may also contribute to inflammation and autoimmunity. Abnormalities of rearrangements at the TCRα locus may also contribute to immune dysregulation in patients with hypomorphic RAG mutations. During normal thymocyte development, multiple waves of V(D)J recombination are required for the expression of distal TRAV genes. Patients with hypomorphic RAG mutations show a dramatic reduction in T cells expressing the products of distal TRAV genes, including TRAV1-2.36 The TRAV1-2 product (Vα7.2) is part of the semi-invariant TCR expressed by human mucosa-associated invariant T cells, which serve an important homeostatic function at mucosal surfaces by recognizing microbial-derived vitamin B metabolism products.37 The reduced number of mucosa-associated invariant T cells may contribute to immune dysregulation at the mucosal surface level and dysbiosis in RAG deficiency.38 Finally, defective V(D)J recombination activity also affects the development of invariant natural killer T cells, another important immunoregulatory subset.34,39 Altogether, abnormalities in T-cell repertoire diversity and composition have important implications for immune deficiency and the immune dysregulation of RAG deficiency. A reduced diversity translates into an impaired capacity to recognize nonself antigens, causing increased susceptibility to infections. On the other hand, qualitative abnormalities in the composition of the T-cell repertoire, with an increased frequency of clonotypes harboring molecular signatures of self-reactivity, represent a risk factor for autoimmune complications.
RAG deficiency also has important consequences for B-cell development and homeostasis. Despite a block at the pre-B cell stage in the bone marrow and a severe reduction in the number of circulating B cells,40 enzyme-linked immunospot analysis showed that Ig-secreting cells are present in the bone marrow of OS patients,41 and autoantibodies are frequently detected, especially in patients with AS and CID-G/ASI, indicating defects in B-cell tolerance checkpoints.26,42 Reexpression of the RAG genes in the bone marrow at a stage between early immature and immature B cells reduces the proportion of autoreactive B cells (a process known as “receptor editing”).43 Impaired receptor editing has been demonstrated in patients and mice with hypomorphic RAG mutations.15,40,41 In the periphery, a second B-cell tolerance checkpoint takes place at the transition between transitional B cells and mature naive B cells. These 2 populations differ in their levels of expression of B-cell activating factor receptor, which serves as a survival signal. Patients and mouse models with hypomorphic RAG mutations are B-cell lymphopenic, a condition that favors the increased B-cell activating factor levels produced by myeloid cells, promoting survival of transitional B cells and enriching the peripheral B pool in self-reactive B cells.15,35 An increased usage of IGHV4-34, IGHV3-9, IGH4-31, and IGHV3-23 genes, which has been associated with autoreactive B cells,44,45 has been reported in RAG-mutated patients.40,46
Patients with OS often present with cutaneous and gastrointestinal (GI) clinical manifestations. The possible mechanisms accounting for immune dysregulation in these compartments have been extensively studied in Rag2R229Q mice, a model of OS. Rag2R229Q mice develop spontaneous signs of intestinal disease, with activated T cells infiltrating the gut lamina propria and expressing high levels of CCR9 (a gut-homing receptor) and T helper 1 (Th1) and Th17 cytokines.38 Lack of secretory IgA, which plays a protective role and acts as a bacterial flora modulator, permits intestinal colonization. Along with mucosal inflammation, increased permeability leads to disruption of the gut mucosa barrier and to bacterial translocation, also documented by increased serum levels of lipopolysaccharide.38 Analysis of the microbiota of Rag2R229Q mice showed a reduction in richness and diversity and enrichment of the Proteobacteria phylum, which is also observed in chronic inflammatory conditions.47 Transfer of fecal samples from wild-type mice into Rag2R229Q mice was associated with decreased inflammation and reduction of cellular infiltrates. In contrast, wild-type mice colonized with Rag2R229Q feces developed increased frequencies of inflammatory cells enriched in Th1/Th17 cells in the gut lamina propria and in the spleen.38 Depletion of microflora using antibiotics (metronidazole, vancomycin, ampicillin) had a dramatic impact on gut inflammation,38 further supporting the notion that dysbiosis plays a causative role in immune dysregulation.48
Finally, a significant proportion of patients with CID-G/AI exhibit extensive granulomas in skin and internal organs.7,8,20,23 The granulomas are characteristically composed of organized inflammatory infiltrates, including centrally localized macrophages and giant cells, a ring of lymphocytes, and variable number of fibroblasts.49 The pathophysiology of granuloma formation remains enigmatic and has not been recapitulated in mouse models carrying mutations equivalent to those identified in patients with CID-G/AI.35 Interestingly, the rubella virus vaccine strain has been detected in granulomatous lesions and other tissue samples of patients with hypomorphic RAG mutations50,51 or with other T-cell defects,52,53 where it establishes chronic infection in M2 macrophages and keratinocytes. It is unknown why only the rubella virus vaccine strain (and not the measles or mumps viruses, for instance) has been identified in the granulomas. Moreover, the observation that granulomas occur at a variable time (often, years) after administration of the rubella vaccine suggests that the immunodeficiency of RAG deficiency plays a critical role in persistence of the attenuated virus, yet additional factors must account for the delayed immunopathology. Mutations of the virus genome and changes in the immune profile of patients (with increased cytokine production associated with ongoing homeostatic proliferation) may play a role in the development of granulomatous lesions.
Immune dysregulation in other forms of CID
In addition to RAG deficiency, immune dysregulation is common in patients with other forms of CID. Thymocyte positive and negative selection, as well as generation of thymic Tregs, are largely controlled by the strength of TCR signaling.54 In particular, weak interaction of the TCR with self-peptide–major histocompatibility complex (MHC) prevents death by neglect and promotes thymocyte survival; in contrast, strong agonist-induced TCR signaling causes negative selection of self-reactive thymocytes55 or induction of CD25+ Treg progenitors.56 Genetically determined defects of TCR signaling are associated with altered T-cell tolerance. For example, autoimmune cytopenias, hypothyroidism, and enteropathy have been reported in patients with CD3γ deficiency,57-59 and the study of their T-cell repertoire has revealed molecular signatures of self-reactivity, with a high proportion of clonotypes containing hydrophobic amino acids, along with a reduced number and restricted repertoire of Tregs.59 Hypomorphic ZAP70 mutations have been associated with wheezing, erythroderma, and inflammatory bowel disease,60,61 and a combination of hypomorphic and activating mutations in the same gene has been reported in 2 siblings with severe bullous pemphigoid, colitis, nephrotic syndrome, and autoantibody-induced hemophilia.62 Autoimmune cytopenias and lymphoproliferative disease were prominent signs of immune dysregulation in 3 siblings with LAT deficiency,63 whereas nodular skin lesions with inflammatory infiltrates, autoimmune thrombocytopenia, and retinal vasculitis have been reported in a patient with LCK deficiency.64
Other forms of CID are due to signaling defects that are not restricted to T cells. The CARD11-BCL10-MALT1 signalosome complex provides a link between triggering of cell surface receptors (including the TCR and the B-cell receptor) to NF-κB signaling.65 Following TCR and B-cell receptor activation, CARD11 is recruited to the immunological synapse, where it is phosphorylated by protein kinase C θ (PKCθ) and PKCβ in T and B cells, respectively.66,67 This allows recruitment of BCL10 and MALT1,68 ultimately leading to NF-κB activation.65 In addition, CARD11 and MALT1 regulate TCR-mediated glutamine uptake and activation of the mTOR pathway.69 Null-CARD11 mutations cause susceptibility to severe infections since early in life, which is associated with an increased proportion of transitional B cells, hypogammaglobulinemia, a predominantly naive T-cell phenotype, severely diminished Treg number, and decreased in vitro T-cell proliferative responses.70 Interestingly, somatic second site reversion allowing partial restoration of CARD11 protein expression and function was associated with an OS-like phenotype, including erythroderma with oligoclonal expansion of skin-infiltrating T cells, hepatosplenomegaly, eosinophilia, and elevated serum IgE.71 In contrast, heterozygous dominant–negative CARD11 mutations are associated with immune dysregulation manifesting as severe atopic disease, associated with Th2 skewing70,72 ; most of these patients also have a history of recurrent respiratory and cutaneous infections, and some also exhibit autoimmunity (colitis, necrotizing granulomas, alopecia).73 In addition to impairing NF-κB activation, these mutations often prevent optimal glutamine uptake in response to TCR stimulation, thereby reducing mTORC1 signaling. MALT1 deficiency is another cause of CID; in addition to infections, these patients also exhibit eczema and inflammatory GI disease, and prominent T-cell infiltrates are often found in skin and GI biopsies.74-76 Interestingly, multiorgan inflammatory disease is also present in Malt1PD/PD mutant mice and is associated with expansion of Th1, Th2, and Th17 cells, high levels of inflammatory cytokines, decreased numbers of IL-10–producing B cells, and a severe defect in Treg number.77
CD40 ligand (CD40L) is a cell surface molecule that is predominantly expressed by activated CD4+ T cells; upon interaction with its cognate receptor CD40, which is constitutively expressed by B cells and antigen-presenting cells, it plays an important role in regulating B-cell responses and promoting T-cell priming.78 In addition, soluble CD40L released by T cells and platelets plays an important role in modulating inflammatory responses.79 CD40L deficiency is an X-linked CID; it is primarily characterized by susceptibility to severe infections that are often caused by opportunistic pathogens, including Pneumocystis jirovecii, Cryptosporidium spp., Cryptococcus, CMV, and Mycobacteria.80 T-cell responses to Cryptosporidium and CMV infection of the liver and bile ducts play an important role in the development of sclerosing cholangitis.81,82 Moreover, ∼7% to 10% of the patients exhibit immune dysregulation, with thrombocytopenia, hemolytic anemia, seronegative arthritis, hypothyroidism, cutaneous granulomas, and inflammatory bowel disease.83-87 These autoimmune complications may reflect, at least in part, skewing of the IgM repertoire toward self-antigens.88 A severe decrease in circulating T follicular cells, Tregs, and follicular regulatory T cells has been reported in 1 patient with autosomal-recessive CD40 deficiency and in adults with CD40L deficiency,89 supporting the concept that the T-cell abnormalities are a major driver of autoimmunity in this disease. Although autoimmune manifestations are also common in B-cell autonomous forms of hyper-IgM syndromes, such as activation-induced cytidine deaminase deficiency,90 the mechanisms are different. In the absence of functional activation-induced cytidine deaminase, central B-cell tolerance and peripheral B-cell tolerance are affected.91,92 The impairment of somatic hypermutation defect causes persistence of autoreactive B-cell clones prevents production of mutated antibodies that can clear antigens. In this setting, immune activation results in sustained production of cytokines that may negatively affect Treg function.91,92
MHC class I and class II molecules play a critical role in adaptive immune responses. By allowing peptide binding and presentation to developing thymocytes, they participate in the mechanisms of positive and negative selection, whereas in the periphery they regulate CD8+ and CD4+ responses. Deficiencies in MHC class I and class II molecules are a cause of CID.1 In addition to suffering from recurrent sinopulmonary infections, patients with MHC class I deficiency have a high propensity to develop sterile necrotizing granulomas that predominantly affect the skin but may also involve the lungs and other tissues.93,94 In 1 case, persistence of the vaccine strain of the rubella virus was demonstrated in the skin lesions.53 Autoreactive natural killer cells and TCRγδ+ T cells have been demonstrated in the circulation and in the skin lesions of these patients,94 suggesting that they may play a role in disease pathophysiology. Use of immunosuppressive medications and injection of IFN-α are not helpful and may worsen the lesions.95 Patients with MHC class II deficiency are highly vulnerable to bacterial, viral, and fungal infections and have a high mortality. Autoimmune manifestations, and cytopenias in particular, are seen in ∼20% of patients.93 Defective MHC class II expression on TECs may cause impaired negative selection of self-reactive CD4+ T cells and contribute to skewing of the CD4+ T-cell repertoire.96,97
Serine threonine kinase 4 (STK4) is a broadly expressed protein upstream of MAPK that controls cell survival and apoptosis. STK4 deficiency in humans is a cause of CID; in addition to increased susceptibility to infections (mostly of viral origin), these patients often exhibit dysgammaglobulinemia with autoantibodies, lymphoproliferative disease, and autoimmune cytopenias, as well as an increased risk for B- and T-cell lymphomas.98-101 It has recently been shown that the mechanisms underlying immune dysregulation in this condition are B-cell autonomous.102 Patients have an increased proportion of transitional B cells and a reduced number of memory B cells, and STK4-deficient mice have a low number of splenic T follicular helper (Tfh) cells, follicular helper B cells, and peritoneal B1a cells, as well as an absence of marginal zone B cells. In response to immunization, Stk4 mutant mice display impaired generation of germinal centers and produce a low number of memory B cells.102 Moreover, although memory B cells from STK4-mutated patients and mice can differentiate into plasma cells, these have an impaired capacity to produce antibodies.102 It is unclear how these B-cell abnormalities are linked to autoimmunity in these patients. One hypothesis is that STK4 deficiency may lead to impaired FOXO1 phosphorylation and nuclear translocation and, consequently, affect germinal center formation and antibody production.102
IL-21 regulates Tfh cell and regulatory B-cell development and promotes plasma blast generation.103,104 IL-21 deficiency is a form of CID described in a patient who presented with very early-onset inflammatory bowel disease and went on to develop recurrent and severe respiratory tract infections.105 The immunological phenotype included hypogammaglobulinemia, a low number of class-switched memory B cells, a normal number of T cells (but with an increased proportion of TCRγδ+ T cells), and reduced T-cell proliferation in vitro to tetanus toxoid. An inflammatory bowel disease–like picture has been also reported in patients with IL-21R deficiency, although in these cases the GI tract pathology may have been driven by Cryptosporidium infection.106,107 Studies in mice have shown that IL-21/IL-21R signaling protects against dextran sulfate sodium–induced colitis through suppression of Th1 responses and activation of Th2 cell, Th17 cell, and Treg responses.108
Inducible T-cell costimulator (ICOS) is a costimulatory molecule that regulates T-cell proliferation and cytokine secretion, migration of Tfh cells into the follicles, and production of IL-10 and IL-21, thereby favoring generation of long-lived memory B cells and plasma cells.109,110 In addition, it has recently been shown that ICOS-mediated costimulation imprints the functional stability of FOXP3 required for optimal Treg function in the periphery.111 Human ICOS deficiency was initially identified as a monogenic form of adult-onset common variable immunodeficiency,112 but it has subsequently been included among the causes of CID.1 The disease is characterized by increased susceptibility to bacterial, viral, and opportunistic infections, immune dysregulation, and cancer.113 The immunological phenotype includes hypogammaglobulinemia, impaired specific antibody responses, a reduced number of class-switched memory B cells and circulating Tfh cells, and defective in vitro production of various cytokines.113 In a series of 15 patients, immune dysregulation was documented in 73% of them, and it presented more commonly as enteropathy; however, splenomegaly, psoriasis, arthritis, cytopenias, eczema, granulomas, and interstitial lymphocytic pneumonitis have been also documented that require the use of steroids or other immunosuppressive agents.113 The precise mechanisms underlying immune dysregulation in this disease are not known. A normal number of Tregs has been reported in most patients,113 but reduced expression of CTLA4 has been observed in some cases.114 Finally, the observation that plasmacytoid dendritic cells promote IL-10 production by Tregs via the ICOS ligand115 suggests that a possible defect of this pathway may also contribute to altered peripheral immune tolerance.
Activated phosphoinositide 3-kinase δ syndrome 1 (APDS1) and APDS2 are due to heterozygous mutations in the PIK3CD or PIK3R1 gene; these encode for the p110δ catalytic and the p85α regulatory subunits of the PI3Kδ enzyme, respectively. The mutations cause increased activity of the PI3K-AKT-mTOR pathway. The clinical phenotypes of APDS1 and APDS2 overlap significantly.116 Although both diseases are listed as “Predominantly antibody deficiencies” in the IUIS classification,1 they are indeed CIDs, with increased susceptibility to sinopulmonary infections, recurrent or persistent infections due to Herpesviridae (Epstein-Barr virus, CMV, varicella zoster virus, herpes simplex virus), lymphoproliferation (lymphadenopathy, splenomegaly and nodular lymphoid hyperplasia in the gut and the respiratory tract), autoimmunity (cytopenias, thyroiditis, glomerulonephritis, type 1 diabetes, autoimmune hepatitis, and others), and a high risk for lymphoma.116 The immunological phenotype of APDS includes (1) T-cell abnormalities with decreased naive T cells, increased T effector memory cells and Tfh cells, and accumulation of senescent T cells, and (2) B-cell impairment, as indicated by variable degrees of hypogammaglobulinemia, elevated IgM levels, increased number of transitional B cells, decreased switched memory cells, and impaired response to immunization.117 Functional testing on T and B cells in APDS patients demonstrated increased AKT and S6 phosphorylation due to increased mTOR signaling, thereby supporting the use of mTOR inhibitors to control the disease.118 HSCT has been successful in reversing the clinical phenotype; however, graft failure and viral reactivation posttransplant have been observed.119,120 Characterization of the molecular mechanisms underpinning APDS prompted the use of selective PI3Kδ inhibitors as a possible therapeutic option. An open-label trial with dose-escalating administration of leniolisib for 12 weeks resulted in significant improvement of lymphoproliferation and cytopenias by the end of the treatment.121
Conclusions
Autoimmunity and lymphoproliferation are increasingly recognized as a primary risk factor for morbidity and mortality in patients with CID. Various mechanisms may contribute to the immune dysregulation that is often observed in patients with CID. The inability to clear infections may result in continuous stimulation of the immune system, increased production of inflammatory cytokines, and tissue damage, as indicated by sclerosing cholangitis following Cryptosporidium or CMV infection in patients with CD40L and CD40 deficiency, as well as by detection of the vaccine rubella virus in granulomas of patients with various forms of CID. However, perturbations in central and peripheral tolerance in T- and/or B-cell compartments also play a major role in causing autoimmunity, hyperinflammatory responses, and increased risk for lymphoproliferation in patients with CID. Improved knowledge of the genetic and molecular mechanisms underpinning immune dysregulation in CID has the potential to provide targeted therapeutic approaches aimed to prevent disease evolution and improve outcome.
Acknowledgment
This work was supported by the Division of Intramural Research, National Institute of Allergy and Infectious Diseases, National Institutes of Health.
Authorship
Contribution: L.D.N., O.M.D., and A.V. researched data and wrote and reviewed the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Luigi D. Notarangelo, Immune Deficiency Genetics Section, Laboratory of Clinical Immunology and Microbiology, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Building 10, Room 5-3950, 10 Center Dr, Bethesda, MD 20892; e-mail: luigi.notarangelo2@nih.gov.