Key Points
STAT6 is a major factor in MF/SS tumorigenesis and a prospective therapeutic target for intervention.
STAT6 promotes proliferation and invasion of MF/SS malignant lymphocytes while progressively depressing the anti-tumor immune response.

Abstract
The signal transducer and activator of transcription 6 (STAT6) is a critical up-stream mediator of interleukin-13 (IL-13) and IL-4 signaling and is constitutively activated in malignant lymphocytes from Sezary syndrome (SS) and mycosis fungoides (MF), the most common subtypes of cutaneous T-cell lymphomas. By combining genome-wide expression profiling with pharmacological STAT6 inhibition, we have identified the genes regulated by STAT6 in MF/SS tumors. We found that STAT6 regulates several common pathways in MF/SS malignant lymphocytes that are associated with control of cell-cycle progression and genomic stability as well as production of Th2 cytokines. Using ex vivo skin explants from cutaneous MF tumors as well as Sezary cells derived from the blood of SS patients, we demonstrated that inhibition of STAT6 activation downregulates cytokine production and induces cell-cycle arrest in MF/SS malignant lymphocytes, inhibiting their proliferation but not their survival. Furthermore, we show that STAT6 promotes the protumoral M2-like phenotype of tumor-associated macrophages in the tumor microenvironment of advanced stage MF by upregulating the expression of genes associated with immunosuppression, chemotaxis, and tumor matrix remodeling. Thus, we show STAT6 to be a major factor in the pathogenesis and progression of MF/SS, promoting proliferation and invasion of the malignant lymphocytes while inducing a progressive depression of the antitumor immune response. Together, our results provide new insights into disease pathogenesis and offer new prospective targets for therapeutic intervention.
Introduction
Cutaneous T-cell lymphomas (CTCL) are a heterogeneous group of lymphoproliferative disorders derived from skin-homing effector memory T cells.1 Mycosis fungoides (MF) and Sezary syndrome (SS) are the most common subtypes.2 MF typically runs an indolent clinical course, characterized by cutaneous patches and plaques infiltrated by malignant T lymphocytes. However, in 20% of patients, the cancer may progress to a fatal advanced stage characterized by tumors, erythroderma, and lymph nodal and visceral involvement.3,4 SS is an aggressive and leukemic form of CTCL characterized by erythroderma, lymphadenopathy, and circulating malignant T cells.5 Diagnosis of MF/SS is difficult, particularly in the early stages, because of the lack of specific markers for malignant lymphocytes, delaying timely treatment. At the advanced stages, treatment options are limited and prognoses are poor.
Aberrant cytokine expression in the MF/SS tumor microenvironment (TME) is a major factor in disease pathogenesis and progression.6-9 Although reactive T helper (Th)1 and CD8+ tumor-infiltrating lymphocytes are found in the TME of early-stage MF/SS, disease progression is accompanied by infiltration with benign and malignant T lymphocytes producing mostly Th2 cytokines (interleukin-4 [IL-4], IL-5, and IL-13).6,7,10-12 In addition, dermal infiltrates of inflammatory cells13-17 and nonimmune resident cells7,9 contribute to the Th2-dominant microenvironment of advanced stage MF/SS, which promotes tumor growth while inducing a progressive immunosuppression underlying the inability of patients to reject tumors and increasing susceptibility to infection.18
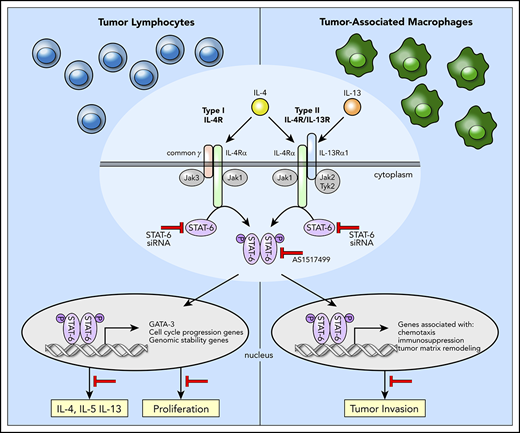
We have previously shown that MF/SS malignant lymphocytes produce high levels of IL-13,19 which acts as an autocrine factor for tumor cells19 and suppresses tumor cell immunosurveillance.11 Furthermore, our studies indicate that IL-13 synergizes with IL-4 in inducing SS cell growth19 and implicate IL-13 signaling via the signal transducer and activator of transcription 6 (STAT6),19 an upstream mediator common to both IL-4 and IL-13 signaling.20 Significantly, we found high numbers of activated STAT6+ cells in the affected skin of MF patients, particularly in advanced stages,19 implicating STAT6 as a critical signaling mediator in MF/SS lymphocytes.
STAT6 is a member of the STAT family of transcription that plays a key role in inflammatory immune responses to IL-4 and IL-1321,22 as well as in tumor immunosurveillance and biology.23-29 Secondary to IL-4/IL-13 stimulation, STAT6 is activated by tyrosine phosphorylation, followed by dimerization and translocation into the nucleus, where it acts as a transcriptional transactivator.20 Although the implication of STAT proteins such as STAT3 and STAT5 in CTCL pathogenesis has been well studied,30-33 little is known of the molecular mechanisms underlying STAT6 regulation of MF/SS pathophysiology.
Combining STAT6 inhibition with genome-wide transcriptome analysis, we identified the STAT6-regulated genes in advanced stage MF/SS tumors. We found that STAT6 inhibition downregulates common molecular pathways in MF/SS malignant lymphocytes which are associated with control of cell-cycle progression, DNA repair, and Th2 cytokine production. Furthermore, inhibition of STAT6 activation downregulates expression of several genes in M2-like tumor-associated macrophages involved with immunosuppression and tumor invasion. These findings provide new insights into MF/SS pathogenesis and offer novel targets for prospective intervention.
Materials and methods
Patient samples
Samples from 28 patients (supplemental Table 1A on the Blood Web site) with confirmed diagnoses of advanced (stage IIB-IVA)2,4,5 MF (n = 8; skin biopsies) and SS (n = 20; blood samples) were obtained from the Comprehensive Skin Cancer Center, Columbia University Medical Center, and from the University of Pittsburgh Medical Center. Healthy control blood (HC, n = 5) and normal skin (NS, n = 8) were obtained from the Central Blood Bank of Pittsburgh and The Health Sciences Tissue Bank, University of Pittsburgh (supplemental Table 1B). Research protocols involving human subjects were approved by the institutional review boards of Columbia University and the University of Pittsburgh. All participants gave written informed consent in accordance with the Declaration of Helsinki.
Cell isolation and culture
CD4+ T cells were isolated from peripheral blood mononuclear cell samples by negative selection using the EasySep Enrichment Kit (StemCell Technologies), as previously reported.34 Cells were isolated from skin using the Whole Dissociation Skin Kitit (Miltenyi Biotec).35 All cultures were performed in complete RPMI-1640 medium (Thermo Fisher Scientific) containing recombinant human IL-2 (10 ng/mL; PeproTech) and IL-7 (5 ng/mL, PeproTech).19 Cells were activated by the Dynabeads CD3/CD28 T-Cell Expander (bead-to-cell ratio: 1:1; Thermo Fisher Scientific). STAT6 was inhibited by the STAT6-specific inhibitor AS151749936 (100 nM; Axon Medchem).
RNA isolation, sequencing, and data analysis
RNA was isolated from samples obtained from SS (n = 4), MF (n = 3), and HC (n = 3) independent donors after 48 hours of culture with/without AS1517499.37 RNA isolation, library preparation, and RNA sequencing followed established procedures detailed in supplemental Methods. Gene ontology analysis was performed using the CLC Genomics Workbench (Qiagen Bioinformatics).
Pathway analysis
Datasets obtained after CLC Genomics Workbench analysis were evaluated by Ingenuity Pathway Analysis (Qiagen). Canonical pathway and upstream regulator analyses were performed on differentially expressed genes (DEGs) as described in Figure 1. In Ingenuity Pathway Analysis, the pathways were compared using a value of P≤ .05.
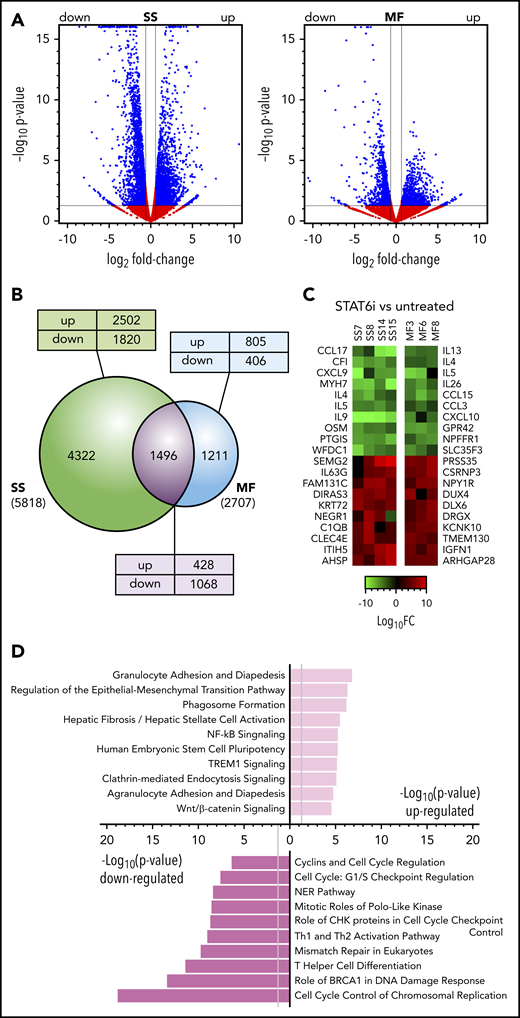
The effect of STAT6 inhibition on transcription in MF and SS tumor samples. (A) Volcano plots comparing gene expression in AS1517499-treated for 48 hours vs untreated MF skin tumors (n = 3) and blood SS cell samples (n = 4). FC, fold change. Blue dots and lines identify the genes scored as differentially expressed (Log2FC > ±0.58 and P < .05). (B) Venn diagram showing the relationship between numbers of DEGs in MF or SS samples after treatment with STAT6 inhibitor or medium alone. The total number of DEGs in SS or MF is given in parenthesis. The number of DEGs unique to SS or MF or shared between them are shown within the diagram; the numbers of genes that were upregulated (up) or downregulated (down) in MF/SS after STAT6 inhibition are also indicated. (C) Heat maps showing a selection of the top DEGs after STAT6 inhibition in individual SS or MF tumor samples. (D) Pathway analysis by Ingenuity of the most significant DEGs commonly up- or downregulated in MF and SS samples after STAT6 inhibition.
The effect of STAT6 inhibition on transcription in MF and SS tumor samples. (A) Volcano plots comparing gene expression in AS1517499-treated for 48 hours vs untreated MF skin tumors (n = 3) and blood SS cell samples (n = 4). FC, fold change. Blue dots and lines identify the genes scored as differentially expressed (Log2FC > ±0.58 and P < .05). (B) Venn diagram showing the relationship between numbers of DEGs in MF or SS samples after treatment with STAT6 inhibitor or medium alone. The total number of DEGs in SS or MF is given in parenthesis. The number of DEGs unique to SS or MF or shared between them are shown within the diagram; the numbers of genes that were upregulated (up) or downregulated (down) in MF/SS after STAT6 inhibition are also indicated. (C) Heat maps showing a selection of the top DEGs after STAT6 inhibition in individual SS or MF tumor samples. (D) Pathway analysis by Ingenuity of the most significant DEGs commonly up- or downregulated in MF and SS samples after STAT6 inhibition.
Single-cell RNA sequencing
Experimental procedures, detailed in supplemental Methods, followed established techniques35 using the Chromium Single Cell 3′ Library V2 Kit (10X Genomics). RNA-sequencing was performed using the Illumina NextSeq500 sequencing system. Cell–gene unique molecular identifier counting matrices generated were analyzed using Seurat.38,39
Intracellular staining of cytokines
Intracellular cytokines staining was performed as previously described34 using the following anti-human antibodies: anti-IL-13, anti-IL-4, anti-IL-5, or respective immunoglobulin G (IgG) isotype controls (all from eBioscience). All samples were collected on an LSRII Flow Cytometer (BD) and analyzed using FlowJo software (BD).
siRNA transfection
Freshly isolated Sezary CD4+ T cells were transfected with 4mM STAT6 ON-TARGETplus SmartPool small interfering RNA (siRNA) or the ON-TARGETplus Non-Targeting siRNA #1 (negative control) (Thermo Fisher Scientific) using the Amaxa Human T Cell Nucleofector kit (Lonza), as previously described.34 STAT6 depletion was determined 5 days after nucleofection by western blot.
Western blotting
Standard methods for immunoblot analysis were used.34 Additional details are reported in supplemental Figure 1.
Cell proliferation and cell metabolic activity
Proliferation of SS cells was determined by carboxyfluorescein diacetate succinimidyl ester assay (CFSE; Thermo Fisher Scientific), according to the manufacturer’s directions. The cellular division capacity was tested by flow cytometry and analyzed by FlowJo. Cell metabolic activity was determined by 3-(4,5-dimethylthiazol-2-yl)-2,5-dimethyltetrazolium bromide; 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) assay (Vybrant MTT Cell Proliferation Kit, Thermo Fisher Scientific) as previously reported.19 Optical density was measured at 570 nm on a Wallac 1420 Victor2 Microplate Reader (PerkinElmer).
Apoptosis assay and cell viability
The Annexin V-FITC Apoptosis Detection Kit (Thermo Fisher Scientific) was used to detect apoptosis of SS CD4+ cells, following the manufacturer's instructions. Samples were analyzed by flow cytometry and FlowJo software. Quantification of cell viability was performed using the Cellometer Auto 2000 Cell Viability Counter (Nexcelom Bioscience), as indicated by the manufacturer.
Cell-cycle analysis
SS CD4+ cells were cultured with/without AS1517499 for 48 hours and stained with Vybrant DyeCycle Ruby Stain Kit (Thermo Fisher Scientific), as indicated. The DNA content was measured by flow cytometry according to the manufacturer's instructions. Analysis of cell-cycle distribution was performed by FlowJo software.
Ex vivo skin explant assays
Organ culture of full-thickness skin explants was performed as previously reported.40,41 Skin punch biopsies (2 × 3 mm) were obtained from MF skin tumors and cultured in 24-well plates in 500 μL complete medium with/without inhibitor. Skin explants were cultured in an air–liquid interface for 5 days, epidermal side up. Cells were fed every 2 days with fresh medium or medium replenished with inhibitor.
Multicolor immunohistochemistry
Multicolor staining was performed on formalin-fixed, paraffin-embedded skin samples using the Tyramide Signal Amplification Kit (Thermo Fisher Scientific) as previously described.35 The antibodies used in these experiments are reported in supplemental Table 3. Confocal images were captured on an Olympus FluoView 1000 Confocal Microscope using an oil immersion 100× objective.
Results
STAT6 inhibition is associated with profound transcriptional changes common to both MF/SS subtypes
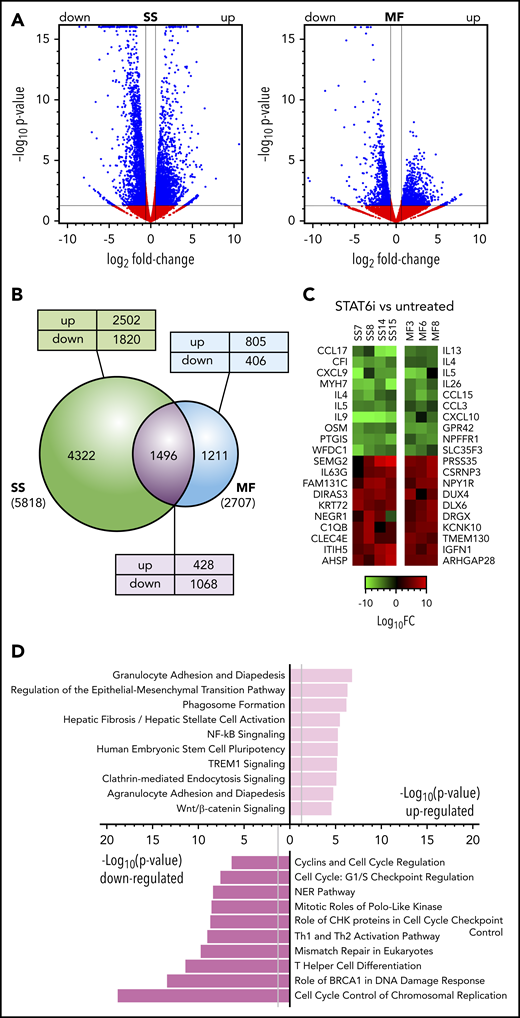
To identify the genes and signaling pathways that are regulated by STAT6 in MF/SS, we investigated the transcriptional changes associated with STAT6 inhibition in primary tumor cells from the blood and skin of advanced stage MF/SS patients. STAT6 was inhibited with AS1517499, a STAT6-specific inhibitor that prevents IL-4-induced STAT6 activation in healthy donor CD4+ T cells (supplemental Figure 1A) while not affecting expression or activation of the upstream Janus kinase (JAK)1, 2, and 3 (supplemental Figure 1B). Pharmacological inhibition of STAT6 resulted in transcriptional changes in all MF/SS samples examined (Figures 1A-B; supplemental Figure 2), although the number of total genes affected in MF samples (n = 2707) was lower compared with SS samples (n = 5818) (Figure 1B). DEG was extremely significant in MF/SS samples after STAT6 inhibition, particularly among those downregulated. Interestingly, 1496 STAT6-regulated genes were shared between MF/SS samples. This unique gene expression signature included 428 upregulated and 1068 downregulated genes (Figure 1B). Heat maps illustrating a selection of the top DEG from individual MF/SS samples tested are shown in Figure 1C. Of note, we found that genes of several cytokines (eg, IL-9, IL-5, IL-4, IL-13, IL-26, OSM) and chemokines (eg, CCL15, CXCL10, CCL17, CCL3, CXCL9) were among the most downregulated in MF/SS samples. Among upregulated DEGs, 2 were tumor suppressor inhibitors (DIRAS3 and ITIH5) found in all SS samples tested, whereas others were specific to either MF or SS samples. To search more broadly for signaling pathways commonly regulated by STAT6 in MF/SS tumors, we performed Ingenuity Pathway Analysis.42 Highly significant examples of shared pathways are shown in Figure 1D. Interestingly, many downregulated pathways were associated with cell-cycle control and progression as well as with DNA damage and repair. STAT6 inhibition also downregulated pathways associated with Th cell differentiation. Upregulated pathways included those associated with inflammation, regulation of epithelial-mesenchymal transition, and Wnt/β-catenin signaling. Altogether, we identified a signature of STAT6-regulated genes common to MF and SS that is associated with distinct cancer-related signaling pathways.
STAT6 inhibition impairs Th2 cytokine production by malignant and reactive MF/SS lymphocytes
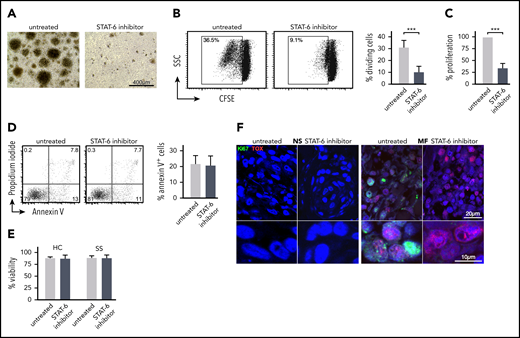
Pharmacological inhibition of STAT6 causes a dramatic downregulation of genes encoding for cytokines such as IL-4, IL-5, and IL-13 (Figure 1C; supplemental Table 2A). We measured protein expression by flow cytometry in peripheral blood SS cells cultured with/without AS1517499 (Figure 2A-B). Contrary to the low transcription level of IL-5, we found extremely high proportions of IL-5+ cells in all samples tested. The proportion of IL-4+ cells was low, in agreement with its transcription level, whereas the number of IL-13+ cells was high, consistent with the transcription level of most samples tested (Figure 2A-B; supplemental Table 2). At the molecular level, we detected a dramatic inhibition of STAT6 activation (Figure 2C; supplemental Figure 1B) and a downregulation of GATA-3 (Figure 2C), the master regulator of Th2 cytokines37 and known target of STAT6.37 Similar results were obtained when STAT6 was silenced by specific siRNAs (Figure 2C; supplemental Figure 3A). By single-cell RNA sequencing (scRNAseq), we show that GATA-3 is highly upregulated in advanced MF tumors compared with NS (Figure 2D; supplemental Figure 4) and that CD3+ lymphocytes and to a lesser extent keratinocytes are the major cell types expressing GATA-3 in MF tumors (Figure 2E). Although GATA-3 expression by human epidermis keratinocytes has been previously described,43,44 GATA-3 expression by MF CD3+ cells likely results from benign and malignant lymphocytes. Indeed, using thymus high-mobility group box (TOX) as a marker of CTCL malignant lymphocytes,19,45,46 we show that GATA-3 is expressed by both TOX− and TOX+ cells, although at a higher level in the latter (P = 1.26 × 10−6; Figure 2F). To evaluate the role of STAT6 on GATA-3 and Th2 cytokine expression by CD3+ lymphocytes in advanced MF tumors, we performed ex vivo skin explant assays from advanced stage tumors cultured for 5 days, with/without inhibitor, followed by multicolor immunofluorescence microscopy. Skin explants maintained intact anatomical structure after culture (Figure 2G). Immunohistochemical staining for GATA-3 revealed numerous CD3+GATA-3+ cells as well as TOX+GATA-3+ cells in untreated tumor explants. Strikingly, STAT6 inhibition completely abolished GATA-3 expression while maintaining unaltered CD3 and TOX expression (Figure 2H-I; supplemental Figure 5). Similarly, IL-4, IL-5, and IL-13 were highly expressed by CD3+ lymphocytes and CD3+TOX+ malignant cells in all the untreated tumor explants tested, whereas STAT6 inhibition completely abolished their expression (Figure 2H-I; supplemental Figure 5). Explants from NS were negative for the expression of GATA-3, Th2 cytokines and TOX. Thus, as an inducer of GATA-3 expression, STAT6 is the main regulator of Th2 cytokines production by MF/SS reactive and malignant lymphocytes.
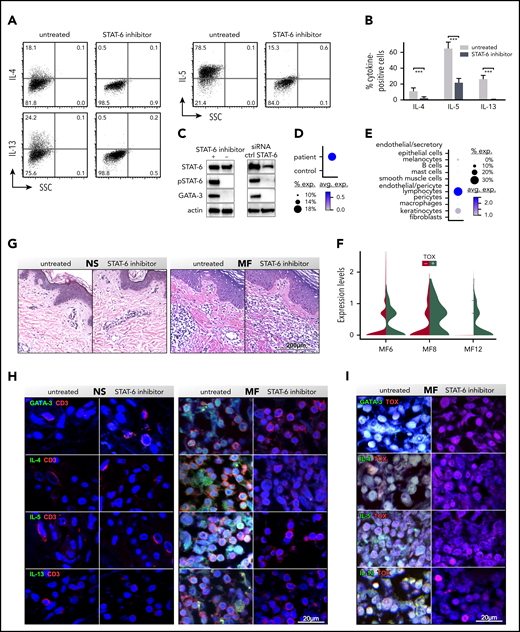
STAT6 inhibition impairs cytokine production by CD3+ MF and SS lymphocytes. SS peripheral blood CD4+ T cells were activated in vitro with CD3/CD28, treated with/without STAT6 inhibitor (AS1517499) for 5 days, and restimulated with PMA and ionomycin. The proportion of IL-4+, IL-5+, and IL-13+ cells was determined by intracellular cytokine staining. (A-B) Representative examples (A) and cytokine frequency (B) shown from multiple SS patients (n = 5, P = .002: Student t test). Mean percentage of positive cells ± standard deviation is shown. (C) Cellular lysates were obtained at day +5 from blood SS CD4+ T cells treated with/without AS1517499 (n = 5) or transfected with STAT6 or target control (crtl) siRNAs (n = 3). Immunoblots were performed with STAT6, phospho (p)STAT6, and GATA-3 antibodies. Actin was used as loading ctrl. (D-E) A representative example is shown. ScRNAseq from fresh MF skin tumors (n = 3; supplemental Figure 4): dot-plots showing the proportion of cells and the scaled average GATA-3 gene expression between MF and NS samples (D) and by different cell types (E). (F) Violin plots show expression of GATA-3 by TOX+ and TOX− lymphocytes from MF tumors. Ex vivo skin explants from the excised MF skin tumors and NS were cultured for 5 days with/without STAT6 inhibition. (G) Hematoxylin and eosin staining, (×200) from representative examples are shown. (H-I) Double-color immunofluorescence staining for Th2 cytokines and CD3 or Th2 cytokines (H) and TOX (I), as indicated (×1000). 4′,6-diamidino-2-phenylindole stains nuclei. Representative NS and MF skin explants out of 5 tumors and 5 controls tested are shown.
STAT6 inhibition impairs cytokine production by CD3+ MF and SS lymphocytes. SS peripheral blood CD4+ T cells were activated in vitro with CD3/CD28, treated with/without STAT6 inhibitor (AS1517499) for 5 days, and restimulated with PMA and ionomycin. The proportion of IL-4+, IL-5+, and IL-13+ cells was determined by intracellular cytokine staining. (A-B) Representative examples (A) and cytokine frequency (B) shown from multiple SS patients (n = 5, P = .002: Student t test). Mean percentage of positive cells ± standard deviation is shown. (C) Cellular lysates were obtained at day +5 from blood SS CD4+ T cells treated with/without AS1517499 (n = 5) or transfected with STAT6 or target control (crtl) siRNAs (n = 3). Immunoblots were performed with STAT6, phospho (p)STAT6, and GATA-3 antibodies. Actin was used as loading ctrl. (D-E) A representative example is shown. ScRNAseq from fresh MF skin tumors (n = 3; supplemental Figure 4): dot-plots showing the proportion of cells and the scaled average GATA-3 gene expression between MF and NS samples (D) and by different cell types (E). (F) Violin plots show expression of GATA-3 by TOX+ and TOX− lymphocytes from MF tumors. Ex vivo skin explants from the excised MF skin tumors and NS were cultured for 5 days with/without STAT6 inhibition. (G) Hematoxylin and eosin staining, (×200) from representative examples are shown. (H-I) Double-color immunofluorescence staining for Th2 cytokines and CD3 or Th2 cytokines (H) and TOX (I), as indicated (×1000). 4′,6-diamidino-2-phenylindole stains nuclei. Representative NS and MF skin explants out of 5 tumors and 5 controls tested are shown.
STAT6 regulates cell-cycle progression in malignant lymphocytes from MF/SS tumors
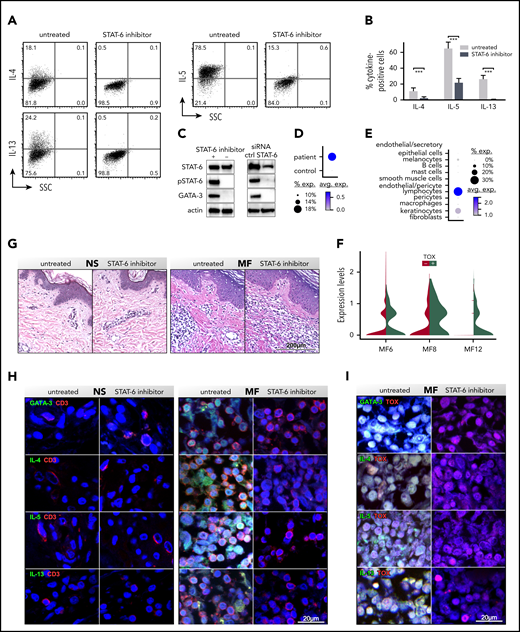
Several molecular pathways associated with cell-cycle progression and genomic stability are downregulated in MF/SS after STAT6 inhibition (Figure 1D). Heat maps in Figures 3A-C depict some representative examples. No similar changes could be detected in HC samples, suggesting that these pathways are specifically activated by STAT6 in MF/SS. In the examples shown, downregulated transcripts included several molecules associated with cell-cycle control of chromosomal replication (Figure 3A), including adenosine triphosphatases (ORC1, ORC6), helicases (CDC6, CDC7, CDC45, mini chromosome region maintenance 2 [MCM2], MCM3 MCM4 MCM5 MCM6, MCM7, MCM8), cyclin-dependent kinases (CDK1, CDK2, CDK4, CDK6), DNA polymerases (POLA1, POLA2, POLD1, POLE, PRIM1, PRIM2), TOP2A, and PCNA. Additional pathways affected by STAT6 inhibition in MF/SS tumor samples included cell-cycle checkpoint control of CHK proteins and of cyclins (Figure 3B-C). In these pathways, downregulated transcripts included regulators of DNA polymerase (RFC1, RFC2, RFC3), PCNA, cyclin-dependent kinases (CDC25A, CDC25C, CDK1, CDK2), and cyclins (CCND2, CCNE2, CCNB2, CCNA2). To functionally validate these findings, we performed cell-cycle analysis in SS CD4+ T cells. Our results (Figure 3D-E) show a significant arrest of cell-cycle progression after STAT6 inhibition as revealed by the increased proportion of cells in G1 phase vs those in G2 phase compared with untreated cells. Single-cell RNAseq demonstrated that the STAT6-regulated genes of Figure 3A-C are overexpressed by TOX+ malignant lymphocytes in advanced MF tumors (Figure 3F-H). We focused on the expression of PCNA, CDK2, and CCNE2 because these genes were highly downregulated after STAT6 inhibition in several cell-cycle regulatory pathways (Figure 3A-C). Multicolor immunofluorescence microscopy on ex vivo skin explants from advanced MF tumors reveals dramatic downregulation of PCNA, CDK2, and CCNE2 protein expression in the explants treated with the STAT6 inhibitor, whereas TOX expression was unaffected (Figures 3I; supplemental Figure 6). Conversely, explants from NS are negative for expression of PCNA, CDK2, CCNE2, and TOX. Together, these data indicate that STAT6 regulates cell-cycle progression in MF/SS malignant lymphocytes.
![STAT6 inhibition induces cell-cycle arrest in MF and SS tumor cells. (A-C) Heat maps showing examples of downregulated pathways after STAT6 inhibition for 48 hours in blood HC (n = 3) and SS (n = 4) CD4+ T cells and MF skin tumor (n = 3) samples. DEGs after STAT6 inhibition are shown in comparison with untreated. (D) Cytometry analysis of the cell cycle of SS CD4+ T cells treated with/without AS1517499 for 48 hours. A representative example showing different phases of the cell cycle. (E) Measured cell-cycle phase distribution shown as a percentage. Data are represented as mean ± standard deviation of 5 independent SS samples (P < .001). (F) Single-cell transcriptomes of 3672 CD3+ cells (256 cells from normal [n = 4] and 3416 cells from MF [n = 3] skin samples, color coded by subject) clustered using Seurat38 (supplemental Figure 4). (G) Transcriptomes of TOX+ T lymphocytes from patient tumors and healthy control skin samples. (H) Dot-plot showing the proportion of cells and the scaled average gene expression of the STAT-6-regulated genes identified in panels A-C in MF and healthy skin samples determined by scRNAseq. (I) Double-color immunofluorescence microscopy of ex vivo skin explants cultured as in Figure 2D-E and stained for PCNA/TOX, CDK2/TOX, and CCNE2/TOX, as indicated, at ×1000. 4′,6-diamidino-2-phenylindole stains nuclei. Representative examples are shown.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/136/15/10.1182_blood.2019004725/2/m_bloodbld2019004725f3.png?Expires=1769376037&Signature=OqqcWZqjAoQppbA9rSy5Bue3D6Dix5aZNvttwCWJoEAJP2JlVcnW4cTI30z2oznDZPntruVzKqlo-mevVDosm0nyw4cEXVe-bFYeE6N810ssC7T1f2zbR3QZ63StLqK4QsozIJomG0Gy55MaclHg3THZ2-SVFEb4nNjMxCQAv5UgKJbWMbIsnhepQ-PBhUECfEU6erRJXGTfCt3r8V0HvJvhp0Xo0gj8VBBf9k4yYCPoJCF6HT0JuniRCPAZ7tQD2sQwuL59gjJR2KQ8k5QFrgdVf2DtHUDwp2lxlSEQ6MphiZF90sMjswC7qgYBLUdAu0O0yDFdzYCiaya27A2jqQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
STAT6 inhibition induces cell-cycle arrest in MF and SS tumor cells. (A-C) Heat maps showing examples of downregulated pathways after STAT6 inhibition for 48 hours in blood HC (n = 3) and SS (n = 4) CD4+ T cells and MF skin tumor (n = 3) samples. DEGs after STAT6 inhibition are shown in comparison with untreated. (D) Cytometry analysis of the cell cycle of SS CD4+ T cells treated with/without AS1517499 for 48 hours. A representative example showing different phases of the cell cycle. (E) Measured cell-cycle phase distribution shown as a percentage. Data are represented as mean ± standard deviation of 5 independent SS samples (P < .001). (F) Single-cell transcriptomes of 3672 CD3+ cells (256 cells from normal [n = 4] and 3416 cells from MF [n = 3] skin samples, color coded by subject) clustered using Seurat38 (supplemental Figure 4). (G) Transcriptomes of TOX+ T lymphocytes from patient tumors and healthy control skin samples. (H) Dot-plot showing the proportion of cells and the scaled average gene expression of the STAT-6-regulated genes identified in panels A-C in MF and healthy skin samples determined by scRNAseq. (I) Double-color immunofluorescence microscopy of ex vivo skin explants cultured as in Figure 2D-E and stained for PCNA/TOX, CDK2/TOX, and CCNE2/TOX, as indicated, at ×1000. 4′,6-diamidino-2-phenylindole stains nuclei. Representative examples are shown.
STAT6 inhibition induces cell-cycle arrest in MF and SS tumor cells. (A-C) Heat maps showing examples of downregulated pathways after STAT6 inhibition for 48 hours in blood HC (n = 3) and SS (n = 4) CD4+ T cells and MF skin tumor (n = 3) samples. DEGs after STAT6 inhibition are shown in comparison with untreated. (D) Cytometry analysis of the cell cycle of SS CD4+ T cells treated with/without AS1517499 for 48 hours. A representative example showing different phases of the cell cycle. (E) Measured cell-cycle phase distribution shown as a percentage. Data are represented as mean ± standard deviation of 5 independent SS samples (P < .001). (F) Single-cell transcriptomes of 3672 CD3+ cells (256 cells from normal [n = 4] and 3416 cells from MF [n = 3] skin samples, color coded by subject) clustered using Seurat38 (supplemental Figure 4). (G) Transcriptomes of TOX+ T lymphocytes from patient tumors and healthy control skin samples. (H) Dot-plot showing the proportion of cells and the scaled average gene expression of the STAT-6-regulated genes identified in panels A-C in MF and healthy skin samples determined by scRNAseq. (I) Double-color immunofluorescence microscopy of ex vivo skin explants cultured as in Figure 2D-E and stained for PCNA/TOX, CDK2/TOX, and CCNE2/TOX, as indicated, at ×1000. 4′,6-diamidino-2-phenylindole stains nuclei. Representative examples are shown.
STAT6 inhibition decreases proliferation of MF/SS malignant lymphocytes
In parallel to the cell-cycle analysis, we investigated the effect of STAT6 inhibition on MF/SS malignant lymphocyte proliferation and survival. We purified CD4+ cells from the peripheral blood of patients with SS and cultured them in vitro with and without AS1517499 for 5 days. We observed that treated cell cultures exhibited consistently decreased frequencies of proliferating cell clusters compared with untreated cultures (Figure 4A). Cell proliferation was assessed by CFSE analysis and MTT assays. Both assays demonstrated a significant decrease in Sezary cell proliferation after STAT6 inhibition (Figure 4B-C). Similar results were obtained when STAT6 was silenced by siRNAs (supplemental Figure 3B). However, we established that AS1517499 treatment did not induce apoptosis in treated cells, as measured by Annexin V analysis (Figure 4D) and did not affect viability of SS or HC CD4+ T cells (Figure 4E), indicating that the inhibitor did not exert any direct cellular toxicity. Ex vivo skin explant assays of MF skin tumors exhibited a decreased proliferation after STAT6 inhibition as indicated by a lower number of Ki67+ cells, a marker of proliferation.47 Dual-staining immunofluorescence microscopy further shows colocalization between Ki67 and TOX, implying inhibition of proliferation in malignant lymphocytes from MF skin tumors (Figure 4F). Thus, STAT6 inhibition affects proliferation but not basal survival of malignant lymphocytes from MF/SS.
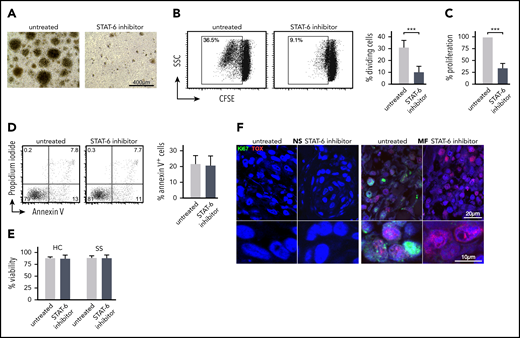
STAT6 regulates proliferation but not survival of malignant lymphocyte in MF and SS patient samples. Peripheral blood SS CD4+ T cells were cultured for 5 days with/without AS1517499. (A) Representative images from the cell culture conditions indicated on day +5. (B) Cell proliferation by CSFE assay. The cellular division capacity was determined by flow cytometry. Gates are set in the dividing cell population. A representative experiment and the mean percentage of divided (CFSElow) cells ± standard deviation (SD) from 5 SS patients are shown. (C) Cell metabolic activity was measured by MTT assay. Results are expressed as percent change of absorbance from untreated control cells, which is set at 100% and represents mean values of triplicate measurements from 11 independent samples. Data are shown as mean ± SD compared with untreated cells. (D) Annexin V and propidium iodide (PI) staining followed by flow cytometry after 48 hours of treatment. Annexin V-positive, PI-negative cells were considered apoptotic. Representative examples and mean percentage of apoptotic cells from 5 SS patients are shown. (E) Cell viability determined by Cellometer Auto 2000 Cell Viability Counter. (F) Representative examples of double-color immunofluorescence microscopy for Ki67/TOX from ex vivo skin explants (MF, n = 5; NS, n = 5) cultured for 5 days with/without STAT6 inhibition as indicated, ×1000. Statistics in panels B-E by Student t test.
STAT6 regulates proliferation but not survival of malignant lymphocyte in MF and SS patient samples. Peripheral blood SS CD4+ T cells were cultured for 5 days with/without AS1517499. (A) Representative images from the cell culture conditions indicated on day +5. (B) Cell proliferation by CSFE assay. The cellular division capacity was determined by flow cytometry. Gates are set in the dividing cell population. A representative experiment and the mean percentage of divided (CFSElow) cells ± standard deviation (SD) from 5 SS patients are shown. (C) Cell metabolic activity was measured by MTT assay. Results are expressed as percent change of absorbance from untreated control cells, which is set at 100% and represents mean values of triplicate measurements from 11 independent samples. Data are shown as mean ± SD compared with untreated cells. (D) Annexin V and propidium iodide (PI) staining followed by flow cytometry after 48 hours of treatment. Annexin V-positive, PI-negative cells were considered apoptotic. Representative examples and mean percentage of apoptotic cells from 5 SS patients are shown. (E) Cell viability determined by Cellometer Auto 2000 Cell Viability Counter. (F) Representative examples of double-color immunofluorescence microscopy for Ki67/TOX from ex vivo skin explants (MF, n = 5; NS, n = 5) cultured for 5 days with/without STAT6 inhibition as indicated, ×1000. Statistics in panels B-E by Student t test.
STAT6 regulates gene expression of tumor-associated macrophages in the TME of advanced stage MF
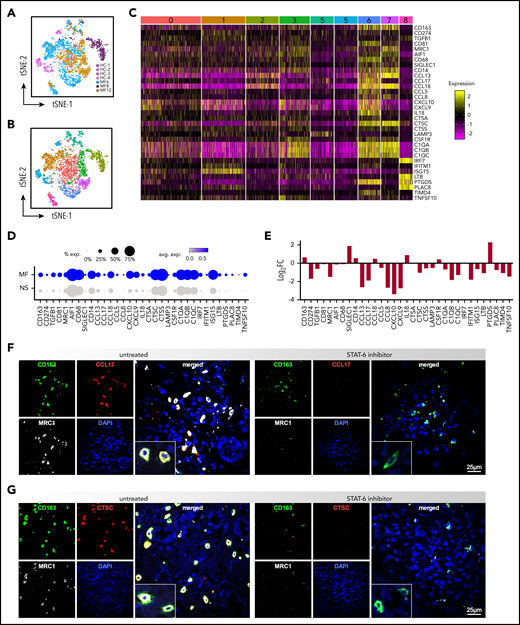
Because STAT6 is a critical signaling mediator of tumor-associated macrophages (TAMs),48,49 we analyzed the effect of STAT6 inhibition on the transcriptome of TAMs from MF skin tumors. ScRNAseq of advanced stage MF samples revealed a high degree of macrophage heterogeneity across patient samples and minimal overlap with the transcriptional profile of macrophages from NS (Figure 5A). Comparison of the macrophage transcriptomes from the tumor and NS samples identified 9 clusters, each exhibiting distinct gene expressions (Figures 5B; supplemental Figure 7A-B). Some macrophage clusters were unique to individual tumors, such as cluster #6 (MF6), #2 (MF8), and #7 (MF12), or controls (cluster #5, HC skin), while clusters #1, 3, 4, and 8 included a mix of macrophages derived from all tumors or tumors and NS samples (cluster #0). Ingenuity Pathway Analysis revealed significant enrichment on pathways such as immune-cell trafficking, cell communication, cytokine signaling, and antigen presentation in tumor-specific macrophage subsets (data not shown). Examples of highly significant DEGs in most of tumor-specific clusters are highlighted by heat map (Figure 5C) and include genes that were previously identified in pro-tumoral M2-like TAMs, including CD163, CD81, MRC1, transforming growth factor β (TGFβ), CCL18, CCL17, CTSS, and CTSC.37,50-54 The proportion of cells and the scaled average expression of these genes by all tumors and controls show strong and specific expression by tumor macrophages (Figure 5D). Significantly, we observed that STAT-6 inhibition downregulated expression of most of the selected genes (eg, CD274, TGFβ, MRC1, C1QB, CCL8, CCL17, CTSC, CXCL10, TIMD4) in MF samples (Figure 5E). We next validated these findings by 3-color immunofluorescence microscopy on MF ex vivo explants. We identified TAMs by expression of CD163 and we determined coexpression with MRC1, CCL17, and CTSC, which are among the top upregulated genes in MF tumor macrophages (Figure 5E). Strikingly, Figure 5F-G shows a strong costain for CD163, MRC1, and CCL17 or CTSC in the untreated patient explants, and shows that STAT-6 inhibition dramatically downregulates expression of CCL17, CTSC and MRC1 but not CD163, in agreement with the gene expression data. Conversely, in NS explants we found only scant CD163+ cells and little to no expression of MRC1, CCL17, and CTSC (supplemental Figure 8). Thus, our findings indicate that STAT6 regulates gene expression of TAMs in the TME of MF patients, favoring immunosuppression and tumor invasion.
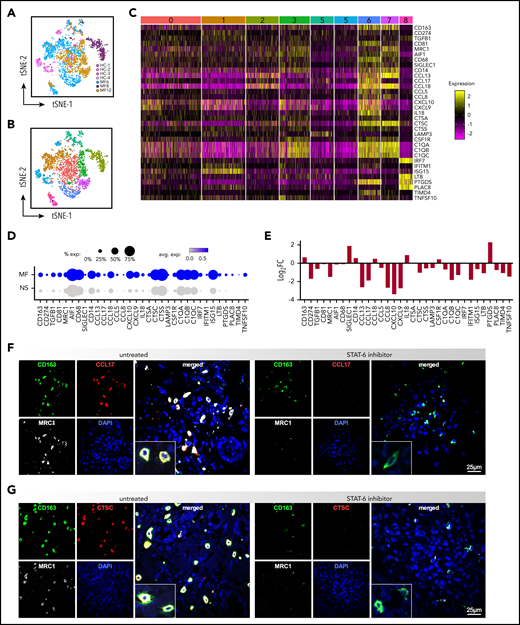
STAT6 regulates gene expression of TAMs in the TME of advanced stage MF. (A) Transcriptomes of macrophages by scRNAseq from individual MF skin tumors (n = 3, 2312 cells) and NS samples (n = 4, 322 cells), color-coded by subject, (B) revealing 9 discrete Louvain clusters. (C) Gene expression from the 9 discrete Louvain clusters in panel B, showing examples of highly significant DEGs in each of the clusters. Cluster numbers are indicated at the top. Each column represents a cell. (D) Dot-plot showing the proportion of cells and the scaled average gene differential expression of the macrophage tumor-associated genes selected in panel C. (E) Bar plot showing the differential expression of the genes selected in panel C after STAT6 inhibition for 48 hours of MF skin tumors (Log2FC > ± 0.58 and P < .05). (F) Three-color immunofluorescence microscopy showing coexpression of CD163, MRC1, and CCL17 or CTSC, in advanced-stage MF skin explants (n = 5) cultured for 5 days with/without AS1517499. Representative examples are shown at ×600, inset double the scale.
STAT6 regulates gene expression of TAMs in the TME of advanced stage MF. (A) Transcriptomes of macrophages by scRNAseq from individual MF skin tumors (n = 3, 2312 cells) and NS samples (n = 4, 322 cells), color-coded by subject, (B) revealing 9 discrete Louvain clusters. (C) Gene expression from the 9 discrete Louvain clusters in panel B, showing examples of highly significant DEGs in each of the clusters. Cluster numbers are indicated at the top. Each column represents a cell. (D) Dot-plot showing the proportion of cells and the scaled average gene differential expression of the macrophage tumor-associated genes selected in panel C. (E) Bar plot showing the differential expression of the genes selected in panel C after STAT6 inhibition for 48 hours of MF skin tumors (Log2FC > ± 0.58 and P < .05). (F) Three-color immunofluorescence microscopy showing coexpression of CD163, MRC1, and CCL17 or CTSC, in advanced-stage MF skin explants (n = 5) cultured for 5 days with/without AS1517499. Representative examples are shown at ×600, inset double the scale.
Discussion
Despite the finding that IL-4/IL-13 signaling via STAT6 plays a critical role in the pathogenesis and progression of MF/SS, the underlying molecular mechanism is still not defined. Combining genome-wide transcriptional profiling with STAT6 inhibition we identified the STAT6-regulated genes in advanced MF/SS tumors. In malignant lymphocytes, we found that STAT6 regulates the expression of genes associated with control of cell-cycle progression, genomic stability, and proliferation. Furthermore, we showed that STAT6 enhances expression of Th2 cytokines in malignant and reactive T cells by upregulating the expression of GATA-3. Finally, we demonstrated that STAT6 contributes to the pro-tumoral M2-like phenotype of TAM in the TME of advanced stage MF by upregulating the expression of genes associated with immunosuppression, chemotaxis, and tumor matrix remodeling. Thus, STAT6 contributes to MF/SS malignancy by several mechanisms including enhancing proliferation of tumor lymphocytes and promoting an immunosuppressive and protumoral microenvironment that favors tumor growth and invasion.
Constitutive STAT6 activation has been observed in several human malignancies.23 Likewise, we found constitutively active STAT6 in MF/SS tumor lymphocytes, particularly in patients with advanced stage tumors.19 In most cancers, STAT6 activation follows IL-4/IL-13 signaling25 ; however, mutations of STAT6 have also been reported but their significance in disease is unclear.28 In B-cell lymphomas, STAT6 is constitutionally activated by JAK2 amplification,55 whereas in Hodgkin lymphoma, its activation depends on the high frequency of genetic lesions in the suppressors of cytokine signaling‐1.56 The predominant mechanism behind persistent STAT6 activation in MF/SS remains to be explored; however, currently available data support the involvement of several potential mechanisms, including activation by increased levels of IL-4 and IL-13 in the TME,6,7 development of an autocrine IL-13 signaling loop,19 as well as cytokine-independent overactivity of JAK1 and JAK3 tyrosine kinases resulting from activating mutation(s).57,58 In preliminary studies, we found that tofacitinib and ruxolitinib (JAK3 and JAK1/2 inhibitors, respectively) decrease proliferation of SS CD4+ T cells in a dose-dependent manner, whereas ruxolitinib but not tofacitinib also downregulates GATA-3 expression (supplemental Figure 9). Interestingly, recent phase 2 clinical trials have shown that the dual SYK/JAK inhibitor, cerdulatinib,59 and ruxolitinib60 demonstrate good tolerability and clinical response in relapsed/refractory CTCL, suggesting that both IL-4 and IL-13 signaling pathways are involved in pathogenesis and make promising therapeutic targets in CTCL.
STAT6 activation contributes to growth, survival, and metastasis of cancer cells in a number of human malignancies.23 Similarly, we demonstrated here that STAT6 promotes proliferation of MF/SS malignant lymphocytes. Mechanistically, we showed that STAT6 regulates expression of a number of cell-cycle regulatory genes during the G1-to-S phase transition, including those involved with cell division and maintenance of genome stability. Transition from G1 to S phase is a critical step during cell division, and its deregulation is a hallmark of cancer.61 Transcriptional changes associated with cell-cycle progression are regulated by specific CDKs and their activating cyclin subunits.62 According to the classical model of cell-cycle control, CCND2 and CDK4 or CDK6 regulate events in early G1 phase, CCNE2–CDK2 triggers S phase, whereas CCNA2–CDK2 and CCNA2–CDK1 are responsible for the completion of S phase. Significantly, we found that STAT6 induces the expression of several CDKs (CDK2, CDK1, CDK4, CDK6) and cyclins (CCNE2, CCND2, CCNA2) in MF/SS malignant lymphocytes, thus committing MF/SS tumor cells to cell division. G1-S CDKs and associated cyclins promote cell-cycle progression mostly by regulating the function of the E2F family of transcription factors, which can act as activators or repressors of transcription.63 Our data indicate that STAT6 inhibition in MF/SS malignant lymphocytes not only downregulates expression of several members of the E2F family (ie, E2F1, E2F7, E2F8), but also abolishes expression of several E2F-regulated genes encoding proteins necessary for initiation of S phase and DNA replication, such as CDC25A, the MCM2-8 family members, CCNA2, and CCNE2. Previous studies reported that STAT-6 itself can specifically recognize a subset of E2F target sites and can regulate cell-cycle gene expression by binding to these DNA elements.64 STAT6 also upregulates expression of other genes involved with DNA replication, including PCNA and components of the DNA polymerase α and δ complexes in malignant MF/SS lymphocytes. Cells rely on DNA structure checkpoints to properly replicate and avoid tumorigenesis by arresting the cell cycle in response to DNA damage or incomplete replication. Our studies indicate that STAT6 regulates several signaling processes associated with maintenance of genome stability in MF/SS, including ATM and CHK pathways as well as responses of BRCA1 in DNA damage and mismatch repair processes. However, although we have established that STAT6 plays a crucial role in regulating cell-cycle progression, other factors such as mutations and epigenetic or posttranslational mechanisms may also contribute to the final outcome of this process.
By upregulating GATA-3 expression, STAT6 induces Th2 cytokine expression by malignant and reactive lymphocytes, promoting the Th2-dominant TME of advanced stage MF/SS. Excess IL-4 and IL-13 in MF local microenvironment induces M2 polarization of TAMs,9,53,65,66 and we found by scRNAseq that TAMs in advanced stage MF samples express several genes previously identified in pro-tumoral CD163+ M2-like TAMs. Among these genes we found a number of chemokines, some of which were previously associated with CTCL.9,13,17,54 Significantly, we established that STAT6 controlled the expression of CCL17, CCL13, and CCL8. Although all 3 chemokines primarily recruit Th2-associated inflammatory cells, they also promote the recruitment of immunosuppressive cells at the tumor site. CCR4, the CCL17 receptor, is selectively expressed on Tregs, Th2 cells, and MF/SS malignant lymphocytes; thus, CCR4-CCL17 interactions are critical for immunosuppression and tumor cell trafficking to MF skin.67 Of note, a recent phase 1/2 clinical trial with mogamulizumab, a humanized anti-CCR4 antibody, showed favorable results in CTCL patients.68 Cancer cells were shown to respond to TAM-synthesized CCL8 by producing CSF1, the major survival and proliferation factor for macrophages, thus promoting an autostimulatory loop.69 In murine models, CCL8 induces tumor cell invasion, motility,70 and metastasis formation.71 Furthermore, CCL8 and CCL13 binding to CCR2, CCR3, and CCR5 recruit Tregs, Th2, monocytes, and eosinophils at the tumor site.72-75 Significantly, expression of CCR3 or CCR4 is associated with a poor prognosis in MF/SS,76 and CCR3 is often expressed by CD30+ large cell transformed tumors in advanced MF/SS.77 STAT6 regulates the immunosuppressive function of MF TAMs by additional mechanisms, including inducing expression of TGF-β, a major suppressor of systemic and local immune responses,78 as well as by stimulating the expression of immune checkpoint receptor ligands such as PD-L1 (also reported as CD274).79 TAMs are also a major source of proteolytic enzymes that degrade the extracellular matrix, favoring the release of matrix-bound growth factors and promoting tumor cell motility and invasion.80 The gene expression profile of TAMs from MF tumors reveals upregulation of genes coding for different matrix proteins (data not shown) as well as several proteolytic enzymes such as cathepsins A, B, C, and S. Previous studies have shown that cathepsin expression at the tumor site is dependent on IL-4 and M2 polarization.81-83 Likewise, we found that STAT6 inhibition abrogates cathepsin expression by TAMs in MF tumors. Additionally, we found that STAT6 induces the expression of several C1q components, including C1qA, B, and C chains by TAMs. Deposits of C1q are found in the stroma and vascular endothelium of several human cancers.84 Significantly, a recent study showed that C1q acts in the tumor microenvironment as a cancer-promoting factor independently of complement activation by favoring adhesion, migration, and proliferation of cancer cells as well as angiogenesis and metastasis.84
We conclude that STAT6 is a major factor in MF/SS malignancy. Targeting STAT6 may represent a new approach to MF/SS treatment, which would potentially accomplish the dual goals of inhibiting proliferation and the invasive potential of malignant lymphocytes, while also enhancing patients’ anti-tumor immunity.
RNA-sequencing data are deposited in Gene Expression Omnibus, available under accession no. GSE147947.
For access to any involved in the study, please contact Patrizia Fuschiotti.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
This work was supported by grants from the National Institutes of Health, National Cancer Institute (R21 CA209107-02) (P.F.) and the Cutaneous Lymphoma Foundation CLARIONS Research Award (P.F.).
Authorship
Contribution: A.M.G. performed experiments and data analysis; L.J.G. contributed to project development; L.J.G., D.S.Q., M.H.T., and O.E.A. acquired samples and collected clinical descriptions; A.M.G. and O.E.A. contributed to manuscript preparation; and P.F. developed the project, performed experiments, analyzed data, and prepared the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Patrizia Fuschiotti, Division of Rheumatology and Clinical Immunology, Department of Medicine, University of Pittsburgh School of Medicine, S709 BST, 200 Lothrop St, Pittsburgh, PA 15261; e-mail: paf23@pitt.edu.

![STAT6 inhibition induces cell-cycle arrest in MF and SS tumor cells. (A-C) Heat maps showing examples of downregulated pathways after STAT6 inhibition for 48 hours in blood HC (n = 3) and SS (n = 4) CD4+ T cells and MF skin tumor (n = 3) samples. DEGs after STAT6 inhibition are shown in comparison with untreated. (D) Cytometry analysis of the cell cycle of SS CD4+ T cells treated with/without AS1517499 for 48 hours. A representative example showing different phases of the cell cycle. (E) Measured cell-cycle phase distribution shown as a percentage. Data are represented as mean ± standard deviation of 5 independent SS samples (P < .001). (F) Single-cell transcriptomes of 3672 CD3+ cells (256 cells from normal [n = 4] and 3416 cells from MF [n = 3] skin samples, color coded by subject) clustered using Seurat38 (supplemental Figure 4). (G) Transcriptomes of TOX+ T lymphocytes from patient tumors and healthy control skin samples. (H) Dot-plot showing the proportion of cells and the scaled average gene expression of the STAT-6-regulated genes identified in panels A-C in MF and healthy skin samples determined by scRNAseq. (I) Double-color immunofluorescence microscopy of ex vivo skin explants cultured as in Figure 2D-E and stained for PCNA/TOX, CDK2/TOX, and CCNE2/TOX, as indicated, at ×1000. 4′,6-diamidino-2-phenylindole stains nuclei. Representative examples are shown.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/136/15/10.1182_blood.2019004725/2/m_bloodbld2019004725f3.png?Expires=1770649205&Signature=gIO2nnnvfDFIRLDXKl9~4R-juzK0dyyBvI~onCCm--zaaaKOBNn6goABwp0wv1-BmG--KJcSX0r12Y3X7d25EQvCRcwzldd5d5wZk40780YtT3wY7wcJoZx29UQ1QcHGWjnduA~w8Ogg8d~J0UWlLqZotoFxveul~wwYCFl-WbHivFeNYSrSCdHskoniwTMp4mf8DmQ673OHXfJMbU11BbwyJwmLhuRfI0Xz8YBshj1c4gMkY3A10HoOCJZOXKvmCCCaBEwFU0fsXbnMw1Hsyck6g-U1FgGjxsb~lLbTVWifz0QY~WHLOYUlDBzYYGpuymmc~ztnUJUfWaMIvIM58A__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)