Key Points
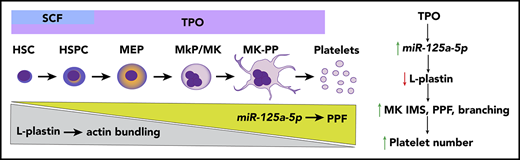
MK miR-125a-5p positively regulates PPF and platelet number by targeting the actin-bundling protein L-plastin.
L-plastin regulates MK migration, PP branching, podosome formation, and invagination membrane system development.

Abstract
There is heritability to interindividual variation in platelet count, and better understanding of the regulating genetic factors may provide insights for thrombopoiesis. MicroRNAs (miRs) regulate gene expression in health and disease, and megakaryocytes (MKs) deficient in miRs have lower platelet counts, but information about the role of miRs in normal human MK and platelet production is limited. Using genome-wide miR profiling, we observed strong correlations among human bone marrow MKs, platelets, and differentiating cord blood–derived MK cultures, and identified MK miR-125a-5p as associated with human platelet number but not leukocyte or hemoglobin levels. Overexpression and knockdown studies showed that miR-125a-5p positively regulated human MK proplatelet (PP) formation in vitro. Inhibition of miR-125a-5p in vivo lowered murine platelet counts. Analyses of MK and platelet transcriptomes identified LCP1 as a miR-125a-5p target. LCP1 encodes the actin-bundling protein, L-plastin, not previously studied in MKs. We show that miR-125a-5p directly targets and reduces expression of MK L-plastin. Overexpression and knockdown studies show that L-plastin promotes MK progenitor migration, but negatively correlates with human platelet count and inhibits MK PP formation (PPF). This work provides the first evidence for the actin-bundling protein, L-plastin, as a regulator of human MK PPF via inhibition of the late-stage MK invagination system, podosome and PPF, and PP branching. We also provide resources of primary and differentiating MK transcriptomes and miRs associated with platelet counts. miR-125a-5p and L-plastin may be relevant targets for increasing in vitro platelet manufacturing and for managing quantitative platelet disorders.
Introduction
There is a growing interest in developing approaches that will enable the in vitro manufacture of a ready, infection-free, and antigen-compatible platelet product,1-5 such that a greater understanding of the molecular mechanisms regulating platelet production is expected to benefit management approaches to thrombocytopenia and thrombocytosis. Platelets are anucleate progeny from mature bone marrow (BM) megakaryocytes (MKs), which in turn are derived from an MK progenitor (MkP) cell.6 Like all cell lineages, MkPs develop from multipotent hematopoietic stem cells (HSCs) that are able to self-renew and proliferate. The classic model of hematopoiesis states that HSCs in the osteoblastic (proliferative) BM niche give rise to independent lineages, including the MK-erythroid progenitor,7 which then differentiates into a separate MkP. Recent evidence also indicates an HSC population with an MK bias.8,9 HSC and MkP expansion is regulated by growth factors, mainly thrombopoietin (TPO), as well as stromal cells and chemokines.10-12 As MKs mature, the cytoplasm fills with a complex network of smooth membrane channels contiguous with the plasma membrane, called the invaginated membrane system (IMS).6,13-15 Mature MKs in the vascular niche undergo transendothelial migration16 and extrude IMS to form long, thin extensions of MKs that contain platelet-sized swellings called proplatelets (PPs).17,18 Platelets or preplatelets are then released into the systemic circulation.11,19 A tubulin-based, dense microtubular cytoskeletal system provides the mechanical forces needed to initiate and elongate PPs.17,18,20 The MK actin cytoskeleton also plays an essential role in platelet production,21 regulating migration, formation of the IMS, and PP branching.6,22,23 Machlus et al have shown that myristoylated alanine-rich C-kinase substrate (MARCKS) regulates late stages of MK PP formation (PPF) and branching,24 but the molecular triggers that initiate PPF from the IMS are not well understood.
Variation in platelet number is heritable25,26 and numerous genetic variants have been associated with platelet count in health and disease.27-29 GATA-1 and FLI1 are transcription factors that play a critical role in stem cell differentiation to early stage MKs, whereas RUNX1 and NFE2 promote MK maturation and PPF.30 Posttranscriptional regulation of gene expression by nonprotein-coding RNAs also regulates megakaryocytopoiesis.31-34 The most heavily studied nonprotein-coding RNAs are microRNAs (miRs), which have a well-defined role in regulating gene expression by interacting with their targeted messenger RNA (mRNA) in a sequence-dependent manner, resulting in inhibition of translation and mRNA degradation.35 Mature, primary murine MKs require protein translation to generate PPs,24 and global knockout of the miR-processing enzyme Dicer results in dysfunctional hematopoietic stem and progenitor cells (HSPCs) with more apoptosis.36 We have shown that MK-specific knockdown of miR-processing ribonucleases Dicer or Drosha causes lower platelet counts.37 However, despite abundant literature linking miRs to murine hematopoiesis and human malignancy, there has been little functional characterizations of miRs and their targets in different stages of normal human megakaryocytopoiesis or thrombopoiesis.31
We have previously used human platelet and MK miR profiling to identify novel genes involved in MK apoptosis and pathways of MK signaling.38,39 The goals of the current study were to identify and characterize miRs and their targets that regulate normal PPF and platelet production. We now report that miR-125a-5p is positively associated with human platelet numbers, and regulates human MK PPF and murine platelet levels. We also demonstrate that miR-125a-5p targets and regulates MK expression of the actin-bundling protein L-plastin (gene name: LCP1); we show that platelet LCP1 transcript levels are negatively associated with healthy human platelet numbers and L-plastin represses human MK PPF, supporting an inhibitory role for actin bundling in PPF. This is the first demonstration of L-plastin in human MKs and defines important regulatory roles for miR-125a-5p and L-plastin in late megakaryocytopoiesis and platelet production.
Methods
RNA profiling of primary human BM MKs, platelets, and cultured MKs
BM aspirates and peripheral blood were obtained during the same encounter from 2 healthy donors. BM MKs were identified based on morphology and staining characteristics (large and polyploid) by a board-certified hematologist, and ∼200 MKs were microdissected by laser-capture scanning microscopy.40 Leukocyte- and erythrocyte-depleted platelets were obtained,41 and human umbilical vein cord blood–derived CD34+ cells were cultured in TPO,39 as previously described39,41 (supplemental Figure 1, available on the Blood Web site). RNA was extracted from immunopurified MKs isolated on days 6, 9, and 13, and 372 common miRs were quantified.
MK PP and branchpoint assessment
An MK PPF assay was performed as described previously39 on in vitro inhibition or overexpression of miR-125a-5p or L-plastin. PP-forming MKs were defined by at least 1 filamentous pseudopod observed by light microscopy at a 20× objective of day 13 cultured cells. Scoring was done blinded as to experimental group. Percentage of PP-forming MKs was the number of MKs with PP divided by the total number of MKs counted times 100%. At least 100 MKs were counted per condition. At least 30 MKs were counted for quantification of PP branchpoints as described.17
In vivo miR-125a-5p LNA inhibition
Mouse studies were performed using 8-week-old C57BL/6 male mice (n = 8 in both groups). miRCURY locked nucleic acid (LNA) inhibitors (22 mg/kg) against miR-125a-5p or negative control A (Exiqon Inc, Hilden, Germany) were administered intraperitoneally every other day over 9 days (5 injections) as previously described.42 On day 10, mice were euthanized, platelets counted, and tissues harvested.
CRISPR/Cas9 knockdown of L-plastin in MKs
Clustered regularly interspaced short palindromic repeats (CRISPR)–CRISPR-associated protein 9 (Cas9) ribonucleoprotein-containing Cas9 nuclease V3 (105 pmol) complexed with LCP1 guide RNA, trans-activating CRISPR RNA, and an electroporation enhancer were transfected by nucleofection into day 3 CD34+ cultures (supplemental Figure 1).
Statistical analysis
Spearman rank correlation (ρ) was calculated for miRs. Pearson correlation (R) was calculated for miR-125a-5p expression levels with platelet count, white blood cells (WBCs), hemoglobin (Hb), or LCP1 levels with platelet count. All other statistical analyses were performed using GraphPad Prism 6 software version 10.1 (La Jolla, CA) and reported as mean plus or minus standard error of mean. The numbers of experiments in the figure legends refer to biologic replicates using different donors, cord bloods, or mice. Significance of in vitro changes in miR-125a-5p and L-plastin levels was determined by 1-sample Student t test. The 1-tailed Mann-Whitney U test was used for calculating in vivo statistical significance. For all other analyses, statistical significance was assessed using the 2-tailed paired Student t test. P < .05 was considered significant.
Results
Transcriptome correlations among primary MKs, platelets, and cultured MKs
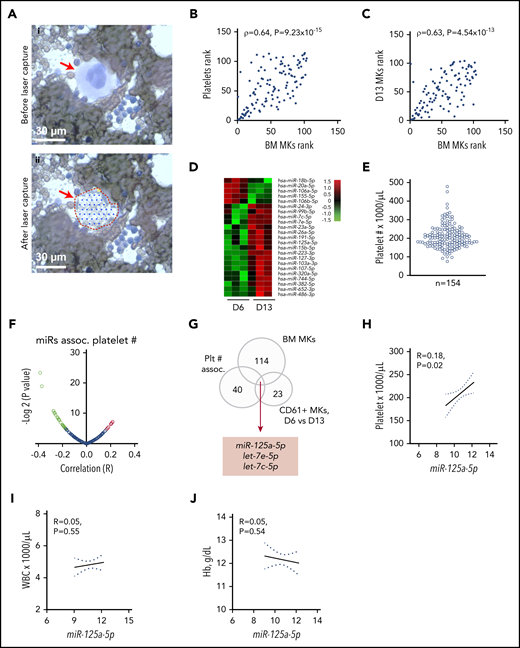
Blood platelet miRs have been associated with numerous clinical and cellular phenotypes. However, it is not known how accurately platelets represent the transcriptome of purified, mature primary BM MKs. To overcome temporal, genetic, and environmental effects, we obtained BM aspirates and peripheral blood from the same healthy donors; total RNA was extracted from mature MKs by laser-capture microdissection (Figure 1A) and from leukocyte- and erythrocyte-depleted platelets.43 Among the 114 miRs shared between platelet and BM MKs, a strong rank correlation was observed (ρ = 0.64; P = 9.23 × 10−15) (Figure 1B; supplemental Table 1 lists the miRs shared by primary platelet and BM MKs).
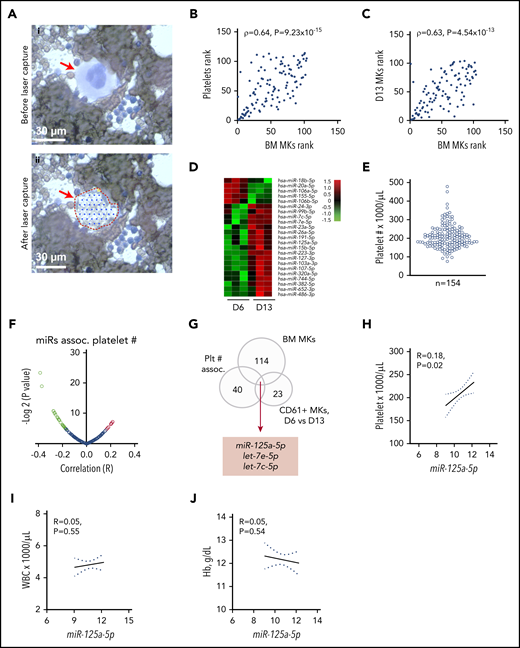
RNA profiling identifies miR-125a-5p association with platelet count. (A) Wright-Giemsa stain of BM aspirate. Scale bar, 30 μm. Same field (i) before and (ii) after laser-capture microdissection (LCM). Red arrow indicates mature MK. (B) Plot of Spearman rank correlation (ρ) of miRs from platelets and LCM BM MKs isolated from the same healthy donors (n = 2). (C) Plot of Spearman rank correlation (ρ) of miRs from CD61-purified, day 13–cultured, cord blood (CB)-derived MKs (n = 3 independent cords) and LCM BM MKs (n = 2 healthy donors). (D) Heat map shows miRs differentially expressed between day 6 (D6) and day 13 (D13) cultured cord blood–derived MKs (n = 3). (E) Variation in platelet count in 154 healthy donors. (F) Volcano plot shows platelet miR association with human platelet count. Green dots indicate negative correlation and red dots shows positive correlation of platelet miRs with platelet count in healthy donors; P < .05. (G) Venn diagram showing intersection of candidate miRs from panels A through F. (H-J) Plots of Pearson correlations (R) of (H) platelet count, (I) WBC, and (J) Hb level against miR-125a-5p levels (n = 154 healthy donors). Dotted lines, 95% confidence intervals. Plt # assoc., platelet number association.
RNA profiling identifies miR-125a-5p association with platelet count. (A) Wright-Giemsa stain of BM aspirate. Scale bar, 30 μm. Same field (i) before and (ii) after laser-capture microdissection (LCM). Red arrow indicates mature MK. (B) Plot of Spearman rank correlation (ρ) of miRs from platelets and LCM BM MKs isolated from the same healthy donors (n = 2). (C) Plot of Spearman rank correlation (ρ) of miRs from CD61-purified, day 13–cultured, cord blood (CB)-derived MKs (n = 3 independent cords) and LCM BM MKs (n = 2 healthy donors). (D) Heat map shows miRs differentially expressed between day 6 (D6) and day 13 (D13) cultured cord blood–derived MKs (n = 3). (E) Variation in platelet count in 154 healthy donors. (F) Volcano plot shows platelet miR association with human platelet count. Green dots indicate negative correlation and red dots shows positive correlation of platelet miRs with platelet count in healthy donors; P < .05. (G) Venn diagram showing intersection of candidate miRs from panels A through F. (H-J) Plots of Pearson correlations (R) of (H) platelet count, (I) WBC, and (J) Hb level against miR-125a-5p levels (n = 154 healthy donors). Dotted lines, 95% confidence intervals. Plt # assoc., platelet number association.
It is also not known how accurately cultured cord blood–derived MKs represent the transcriptome of purified primary BM MKs. Culture conditions using TPO showed the appearance of mature MK markers at day 9,39 and thereafter a steady increase in PPF was observed, peaking at day 13 to 14, similar to work by Balduini et al.44 We immunopurified MKs at day 6, 9, and 13 cultures, extracted RNA, and profiled miRs (see supplemental Table 2 for miRs detected in differentiating MK cultures). Day 13 MKs were taken to be most representative of primary BM MKs based on the highest percentage of mature MK markers and polyploidy.39 Figure 1C shows the correlation between day 13 cord blood–derived CD61+ MKs and laser-captured primary BM MKs miRs (ρ = 0.63, P = 4.54 × 10−13). These data support the rationale for cord blood–derived MKs as a model for primary BM MK gene expression.
miR-125a-5p regulates thrombopoiesis
We hypothesized that changes in gene expression occurring during MK differentiation would contribute to thrombopoiesis, and identified 23 cord blood–derived MK miRs with significantly altered expression during MK maturation and PPF (Figure 1D). To minimize false-positive candidates for testing in CB MKs, we considered only: (1) the 40 miRs that were associated with platelet count in our data set of healthy human subjects45 (Figure 1E-F; supplemental Table 3 contains platelet miRs associated with platelet count); (2) miRs expressed in primary BM MKs (Figure 1A; supplemental Table 1); and (3) the 23 miRs that significantly changed during MK differentiation (Figure 1D). The intersection of these sets of miRs (Figure 1G) consisted of miR-125a-5p, let-7e-5p, and let-7c-5p. Subsequent studies focused on miR-125a-5p because of prior work showing its importance in murine hematopoiesis.46-49 Importantly, miR-125a-5p levels correlated positively with platelet counts, but not with WBC counts or Hb (Figure 1H-J), suggesting a lineage-preferential role. miR-125a-5p has not been studied in normal human megakaryocytopoiesis, PPF, or platelet production.
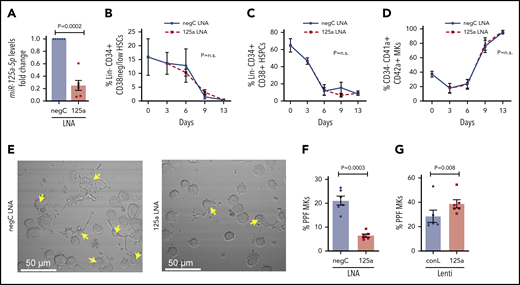
When miR-125a-5p was inhibited by ∼75% in cultures promoting unilineage MK differentiation (Figure 2A), there was no effect on the expected decrease of Lin−CD34+CD38−/low HSCs or Lin−CD34+CD38+ HSPCs (Figure 2B-C), or the increase in CD34−CD41a+CD42a+ mature MKs (Figure 2D). These results suggested that miR-125a-5p had no effect on MK maturation, but the association with platelet number raised the possibility of a miR-125a-5p effect on late-stage MK maturation and thrombopoiesis. Indeed, miR-125a-5p inhibition induced a 70% reduction in PPF (Figure 2E-F; see supplemental Figure 2 for MK tubulin stain). Furthermore, lentiviral-mediated overexpression of miR-125a-5p induced an approximately fivefold increase in MK miR-125a-5p (supplemental Figure 3) and increased MK PPF by ∼35% (Figure 2G).
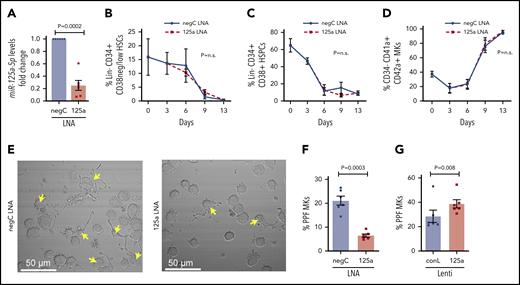
miR-125a-5p regulates MK PPF. (A-F) LNAs were used to inhibit miR-125a-5p (“125a LNA” throughout figure). (A) Fold change of miR-125a-5p compared with control after LNA inhibition (n = 6). (B-D) Flow cytometric analysis of the effect of miR-125a-5p inhibition on (B) HSCs, (C) HSPCs, and (D) mature MKs at days 0, 3, 6, 9, and 13 (n = 3). (E) Representative brightfield images of MKs with PPs (arrows) after miR-125a-5p inhibition. Scale bar, 50 μm. (F) Plot of percentages of day 13 cells with PPs after treatment with LNA inhibitors, scored blinded as to the treatment group (n = 6). (G) Plot of percentages of day 13 cells with PPs after transduction with miR-125a-5p overexpressing lentivirus. conL lenti, a mutated sequence of miR-125a-5p lentiviral plasmid used for transduction assays as a negative control (n = 6); negC LNA, negative LNA control; n.s., not significant.
miR-125a-5p regulates MK PPF. (A-F) LNAs were used to inhibit miR-125a-5p (“125a LNA” throughout figure). (A) Fold change of miR-125a-5p compared with control after LNA inhibition (n = 6). (B-D) Flow cytometric analysis of the effect of miR-125a-5p inhibition on (B) HSCs, (C) HSPCs, and (D) mature MKs at days 0, 3, 6, 9, and 13 (n = 3). (E) Representative brightfield images of MKs with PPs (arrows) after miR-125a-5p inhibition. Scale bar, 50 μm. (F) Plot of percentages of day 13 cells with PPs after treatment with LNA inhibitors, scored blinded as to the treatment group (n = 6). (G) Plot of percentages of day 13 cells with PPs after transduction with miR-125a-5p overexpressing lentivirus. conL lenti, a mutated sequence of miR-125a-5p lentiviral plasmid used for transduction assays as a negative control (n = 6); negC LNA, negative LNA control; n.s., not significant.
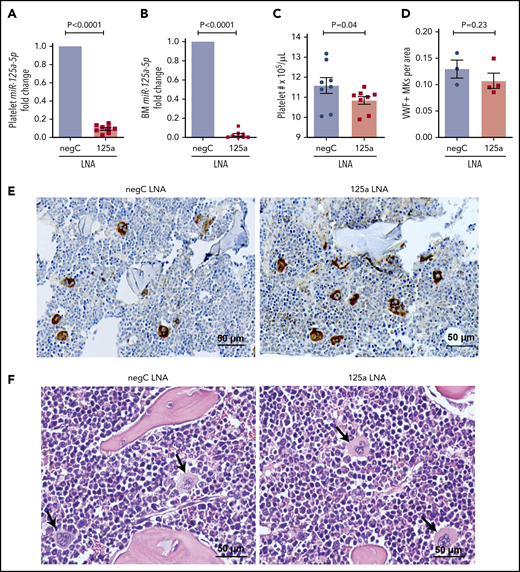
Because MK PPF is believed to be a necessary step for the generation of peripheral blood platelets, we tested whether the in vivo inhibition of miR-125a-5p would alter wild-type (WT) mouse platelet numbers. Compared with control mice, platelets from mice receiving anti-miR to miR-125a-5p had reduced levels of platelet and BM miR-125a-5p (Figure 3A-B), and the platelet counts were modestly but significantly lower (Figure 3C). There was no obvious effect of miR-125a-5p inhibition on BM MK number (Figure 3D-E) or morphology (Figure 3F). There was a nonsignificant trend for decrease in the average spleen weight in the miR-125a-5p–inhibited mice (supplemental Figure 4A), but spleen and liver histology were normal (supplemental Figure 4B-C). Together with the human platelet associations and the in vitro MK studies, the murine antago-miR experiment is consistent with miR-125a-5p as a positive regulator of MK PPF and peripheral blood platelet counts.
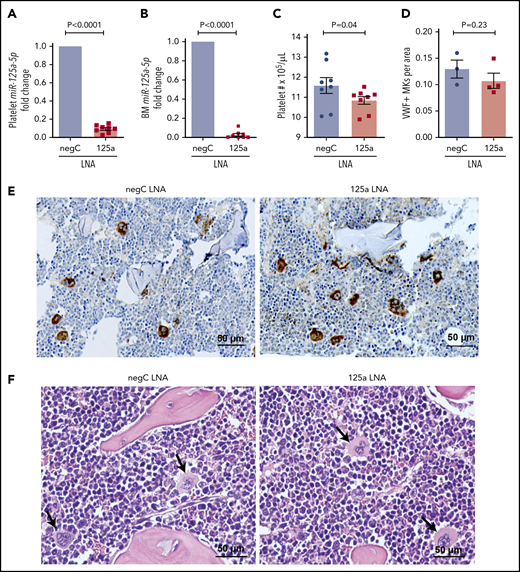
miR-125a-5p regulates platelet count in vivo. LNAs to miR-125a-5p (125a LNA) or negative LNA control (negC LNA) were administered intraperitoneally to C57BL/6 WT mice every other day for 9 days. On day 10, mice were euthanized and assays performed. Fold changes in murine platelet (A) and BM (B) miR-125a-5p levels compared with negC LNA control (n = 8 in each group). (C) Platelet counts from both groups of mice (n = 8). (D) Quantification of von Willebrand factor (VWF)-positive MKs in BM of mice femurs per area (scored blinded as to the treatment group) (n = 3 for negC LNA and n = 4 for 125a LNA). (E) Representative image of VWF immunohistochemical staining of BM (n = 4 in each group). (F) Representative hematoxylin-and-eosin staining of BM (n = 4 in each group). Arrows indicate MK. (E-F) Scale bar, 50 μm.
miR-125a-5p regulates platelet count in vivo. LNAs to miR-125a-5p (125a LNA) or negative LNA control (negC LNA) were administered intraperitoneally to C57BL/6 WT mice every other day for 9 days. On day 10, mice were euthanized and assays performed. Fold changes in murine platelet (A) and BM (B) miR-125a-5p levels compared with negC LNA control (n = 8 in each group). (C) Platelet counts from both groups of mice (n = 8). (D) Quantification of von Willebrand factor (VWF)-positive MKs in BM of mice femurs per area (scored blinded as to the treatment group) (n = 3 for negC LNA and n = 4 for 125a LNA). (E) Representative image of VWF immunohistochemical staining of BM (n = 4 in each group). (F) Representative hematoxylin-and-eosin staining of BM (n = 4 in each group). Arrows indicate MK. (E-F) Scale bar, 50 μm.
miR-125a-5p targets and regulates MK expression of the actin-bundling protein L-plastin
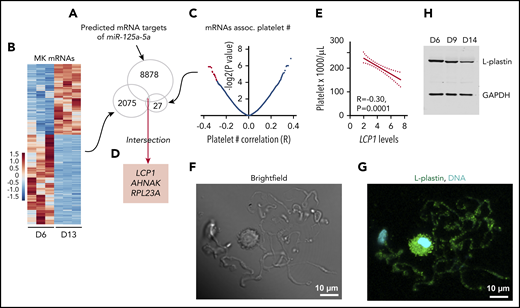
To consider how an increasing level of miR-125a-5p during MK maturation might exert its positive effects on platelet production, we determined the in silico predicted mRNAs that were (1) targeted by miR-125a-5p (Figure 4A); (2) decreased during day 6 to 13 cultured MK differentiation, since miR-125a-5p increased (Figure 4B; supplemental Table 4 lists mRNAs expressed at day 6, 9 and 13 of cultured MKs); and (3) associated negatively with platelet number in healthy subjects (Figure 4C). The intersection of these mRNAs was LCP1, AHNAK, and RPL23A (Figure 4D). We focused on LCP1 because its product, L-plastin, is a hematopoietic-specific actin-bundling protein that regulates T-cell development and migration,50 but has not been studied in MKs. Figure 4E shows the negative association between human platelet LCP1 mRNA levels and platelet count. L-plastin protein was detected in MK cytoplasm and in PPs themselves (Figure 4F-G; see supplemental Figure 5 for immunoglobulin G control). L-plastin mRNA and protein levels decreased from day 6 to 14 in cultures (Figure 4H; supplemental Figure 6), consistent with the inverse relationship with miR-125a-5p during MK differentiation (supplemental Figure 6).
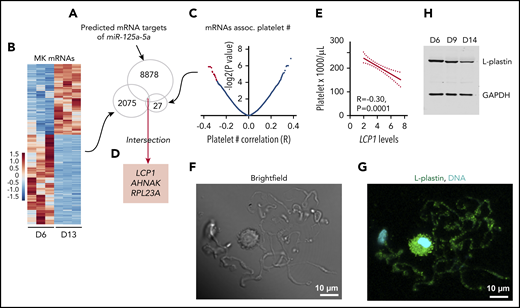
L-plastin identified as a candidate target of miR-125a-5p. (A) Predicted mRNA targets of miR-125a-5p were identified (total, 8878). (B) Heat map displaying differential gene-expression patterns of CD61-purified cultured MKs during differentiation for day 6 (D6) vs day 13 (D13) (n = 3 independent cords). Only mRNAs that showed downregulation during MK differentiation (total, 2075) were further considered. (C) Volcano plot shows platelet mRNA association with human platelet count. Only 27 mRNAs (red dots, some data points are superimposed) that were negatively associated with platelet count (adjusted P < .05) were considered. (D) Intersection of candidate targets from panels A-C. (E) Plot of Pearson correlation (R) between platelet count and LCP1 mRNA levels. Dotted lines, 95% confidence intervals. (F-G) Representative images of day 14 cultured MKs. Scale bar, 10 μm. (F) Brightfield and (G) L-plastin (green) and 4′,6-diamidino-2-phenylindole (DAPI), a nuclear stain (cyan) in round MK and PPs. (H) Representative immunoblot shows L-plastin protein levels in CD34+ cord blood–derived MK cultures at days 6, 9, and 14. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
L-plastin identified as a candidate target of miR-125a-5p. (A) Predicted mRNA targets of miR-125a-5p were identified (total, 8878). (B) Heat map displaying differential gene-expression patterns of CD61-purified cultured MKs during differentiation for day 6 (D6) vs day 13 (D13) (n = 3 independent cords). Only mRNAs that showed downregulation during MK differentiation (total, 2075) were further considered. (C) Volcano plot shows platelet mRNA association with human platelet count. Only 27 mRNAs (red dots, some data points are superimposed) that were negatively associated with platelet count (adjusted P < .05) were considered. (D) Intersection of candidate targets from panels A-C. (E) Plot of Pearson correlation (R) between platelet count and LCP1 mRNA levels. Dotted lines, 95% confidence intervals. (F-G) Representative images of day 14 cultured MKs. Scale bar, 10 μm. (F) Brightfield and (G) L-plastin (green) and 4′,6-diamidino-2-phenylindole (DAPI), a nuclear stain (cyan) in round MK and PPs. (H) Representative immunoblot shows L-plastin protein levels in CD34+ cord blood–derived MK cultures at days 6, 9, and 14. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
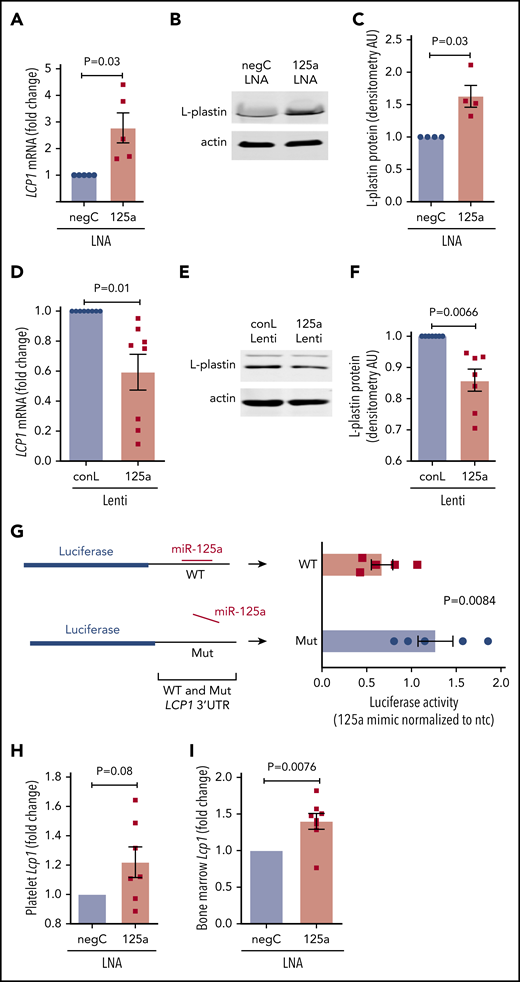
miR-125a-5p is predicted to target the 3′ untranslated region (3′ UTR) of LCP1 (supplemental Figure 7). miR-125a-5p inhibition in cultured MKs induced derepression (ie, increase) of LCP1 mRNA (Figure 5A) and protein (Figure 5B-C). Lentiviral overexpression of miR-125a-5p in cultured MKs caused a significant reduction in LCP1 mRNA (Figure 5D) and protein (Figure 5E-F; supplemental Figure 8). Consistent with an miR-mRNA functional complex, MK argonaute 2 (Ago 2) immunoprecipitations were enriched in both miR-125a-5p and LCP1 mRNA (supplemental Figure 9). Cotransfection of an miR-125a-5p mimic with reporter constructs containing either a WT or mutant LCP1 3′ UTR sequence demonstrated that miR-125a-5p directly targets the in silico predicted LCP1 3′ UTR-binding sequence (Figure 5G). Of note, LCP1 mRNA levels were enhanced in platelets and BM of the mice receiving in vivo anti–miR-125a (Figure 5H-I). Taken together, these inhibition and overexpression experiments demonstrate that miR-125a-5p regulates MK L-plastin levels.
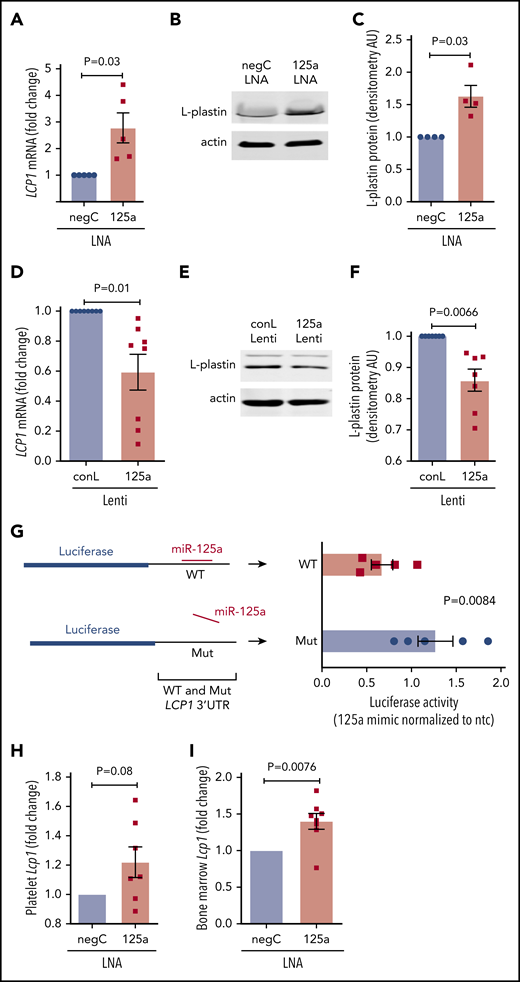
miR-125a-5p directly regulates L-plastin expression. (A) Real-time quantitative polymerase chain reaction (qPCR) quantification of MK LCP1 mRNA after LNA inhibition of miR-125a-5p (125a LNA) shown as fold change vs negative LNA control (negC) (n = 5). (B) Representative immunoblot of MK L-plastin after miR-125a-5p inhibition. (C) Fold changes of densitometric quantification of L-plastin immunoblots after miR-125a-5p inhibition vs negC, normalized to actin (n = 4). (D) Real-time qPCR quantification of MK LCP1 mRNA fold change after miR-125a-5p (125a Lenti) or control (conL) lentiviral overexpression (n = 8). (E) Representative immunoblot of MK L-plastin after miR-125a-5p overexpression. (F) Fold changes of densitometric quantification of L-plastin immunoblots after miR-125a-5p vs conL overexpression, normalized to actin (n = 7). (G) Luciferase reporter constructs with WT or mutated (Mut) 3′ UTR of LCP1 (left) were cotransfected into HEK293T with a pre-miR precursor of miR-125a-5p (mimic) or a nontargeting miR negative control (ntc) and assayed for luciferase activity (n = 5). (H-I) In vivo murine Lcp1 mRNA levels were assessed by real-time qPCR from mice injected with LNAs as in Figure 3. Fold changes are shown for miR-125a-5p vs negC LNA groups. (H) Purified mouse platelets (n = 7). (I) BM from mouse femurs (n = 8). AU, arbitrary unit.
miR-125a-5p directly regulates L-plastin expression. (A) Real-time quantitative polymerase chain reaction (qPCR) quantification of MK LCP1 mRNA after LNA inhibition of miR-125a-5p (125a LNA) shown as fold change vs negative LNA control (negC) (n = 5). (B) Representative immunoblot of MK L-plastin after miR-125a-5p inhibition. (C) Fold changes of densitometric quantification of L-plastin immunoblots after miR-125a-5p inhibition vs negC, normalized to actin (n = 4). (D) Real-time qPCR quantification of MK LCP1 mRNA fold change after miR-125a-5p (125a Lenti) or control (conL) lentiviral overexpression (n = 8). (E) Representative immunoblot of MK L-plastin after miR-125a-5p overexpression. (F) Fold changes of densitometric quantification of L-plastin immunoblots after miR-125a-5p vs conL overexpression, normalized to actin (n = 7). (G) Luciferase reporter constructs with WT or mutated (Mut) 3′ UTR of LCP1 (left) were cotransfected into HEK293T with a pre-miR precursor of miR-125a-5p (mimic) or a nontargeting miR negative control (ntc) and assayed for luciferase activity (n = 5). (H-I) In vivo murine Lcp1 mRNA levels were assessed by real-time qPCR from mice injected with LNAs as in Figure 3. Fold changes are shown for miR-125a-5p vs negC LNA groups. (H) Purified mouse platelets (n = 7). (I) BM from mouse femurs (n = 8). AU, arbitrary unit.
L-plastin regulates MK PPF and branching
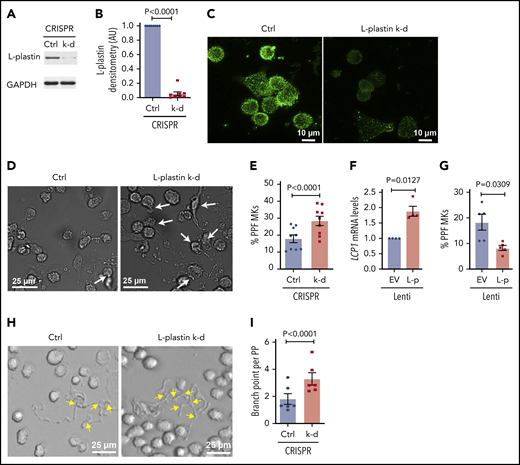
CRISPR-Cas9 gene editing was used to efficiently knockdown L-plastin in primary human MKs (Figure 6A-C), resulting in enhanced MK PPF (Figure 6D-E), consistent with the inverse relationship with platelet number (Figure 4E), and suggesting that L-plastin inhibits platelet production. A similar increase in MK PPF was observed when L-plastin was knocked down with LNA GapmeRs (supplemental Figure 10). Overexpression of L-plastin in MKs (Figure 6F) showed reduction in MK PPF (Figure 6G). PP branching is a mechanism that enables production of thousands of platelets per MK, and L-plastin knockdown significantly increased PP branch points (Figure 6H-I). These data indicate that L-plastin has a negative regulatory role in PPF and branching.
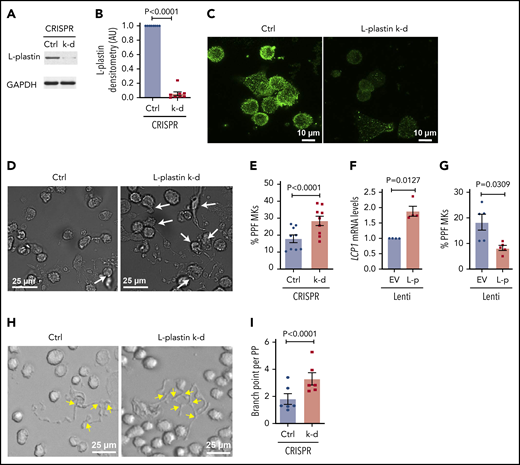
L-plastin regulates MK PPF and branching. (A) Representative immunoblot of L-plastin after CRISPR-Cas9 knockdown (k-d) in day 10 human CD34+-derived cultured MKs. Guide RNAs not targeting known genes are used as negative control (Ctrl). (B) Fold changes of MK L-plastin knockdown assessed by densitometry (n = 8). (C) Ctrl and CRISPR L-plastin k-d MKs on day 14 were stained with anti–L-plastin antibody (green) and imaged using a confocal microscope (60× objective). Scale bar, 10 μm. Staining for both Ctrl and k-d was performed under identical conditions and fluorescence acquisition for both images was obtained with identical microscope settings. (D) Representative brightfield images of cultured PPF MKs (white arrows) after L-plastin knockdown. Scale bar, 25 μm. (E) The percentage of MKs with PPs, scored blinded, after L-plastin knockdown (n = 9). (F) Fold changes of LCP1 expression assessed by real-time qPCR after L-plastin (L-p Lenti) or empty vector (EV Lenti) lentiviral overexpression (n = 4). (G) The percentage of MKs with PPs after L-plastin overexpression scored blinded as to the treatment group (n = 5). (E,G) At least 100 MKs per condition per cord were analyzed. (H) Brightfield image shows MK PP branches (yellow arrows) after L-plastin knockdown. Scale bar, 25 μm. (I) Quantification of MK PP branching, blinded to the treatment groups. At least 30 MKs per condition per cord were analyzed for n = 6 independent cultures.
L-plastin regulates MK PPF and branching. (A) Representative immunoblot of L-plastin after CRISPR-Cas9 knockdown (k-d) in day 10 human CD34+-derived cultured MKs. Guide RNAs not targeting known genes are used as negative control (Ctrl). (B) Fold changes of MK L-plastin knockdown assessed by densitometry (n = 8). (C) Ctrl and CRISPR L-plastin k-d MKs on day 14 were stained with anti–L-plastin antibody (green) and imaged using a confocal microscope (60× objective). Scale bar, 10 μm. Staining for both Ctrl and k-d was performed under identical conditions and fluorescence acquisition for both images was obtained with identical microscope settings. (D) Representative brightfield images of cultured PPF MKs (white arrows) after L-plastin knockdown. Scale bar, 25 μm. (E) The percentage of MKs with PPs, scored blinded, after L-plastin knockdown (n = 9). (F) Fold changes of LCP1 expression assessed by real-time qPCR after L-plastin (L-p Lenti) or empty vector (EV Lenti) lentiviral overexpression (n = 4). (G) The percentage of MKs with PPs after L-plastin overexpression scored blinded as to the treatment group (n = 5). (E,G) At least 100 MKs per condition per cord were analyzed. (H) Brightfield image shows MK PP branches (yellow arrows) after L-plastin knockdown. Scale bar, 25 μm. (I) Quantification of MK PP branching, blinded to the treatment groups. At least 30 MKs per condition per cord were analyzed for n = 6 independent cultures.
L-plastin regulates MK cellular features critical for MK migration and PPF
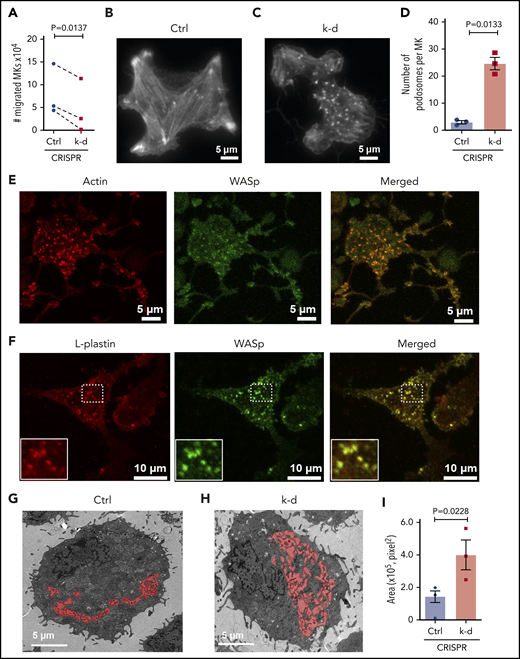
Earlier stages of megakaryocytopoieis are believed to require migration from the proliferative niche to the vascular niche,7 and L-plastin knockdown also caused a significant reduction in MK migration (Figure 7A). To consider whether the actin-bundling functions of L-plastin are involved in MK PPF, filamentous actin (F-actin) was analyzed by total internal reflection fluorescence microscopy. Assembly of the actin-based stress fibers needed for migration was also inhibited by MK L-plastin knockdown (Figure 7B-C). These data indicate that L-plastin promotes MK migration, similar to its effects on lymphocyte migration.51,52
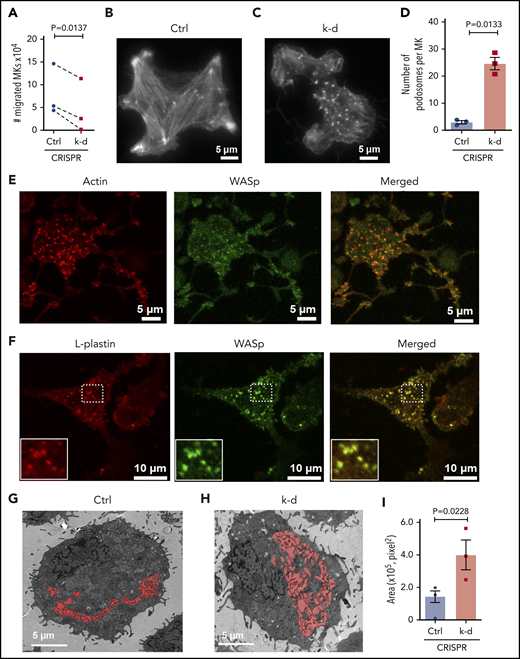
L-plastin regulates MK migration, podosome formation, and IMS. (A) MK migration in transwells toward the SDF-1α chemoattractant on day 13. CD41a+CD42a+ MKs quantified by flow cytometry and normalized to no SDF-1α (n = 3). (B-C) Representative F-actin staining of spread MKs on day 11 by total internal reflection fluorescence (TIRF) microscopy for (B) control MKs and (C) MKs with L-plastin knockdown. (D) Quantification of MK podosomes per MK without or with L-plastin knockdown on day 11 by TIRF (n = 3). At least 25 MKs per condition per cord were analyzed. (E) Representative image of day 14 cultured MKs were stained for actin using ActinRed 555 ready probes (shown in red) and Wiskott-Aldrich syndrome protein (WASp) using anti-WASP antibody Alexa Fluor 488 (shown in green). Scale bar, 5 μm. (F) Representative image of day 14 cultured MKs on fibrinogen were stained for L-plastin using anti-L-plastin primary antibody and Alexa Fluor 555 seconday antibody (shown in red) and WASp stained with anti-WASP primary antibody and Alexa Fluor 488 seconday antibody (shown in green). White box shows magnified image. Scale bar, 10 μm. (G-H) Representative transmission electron microscopic images showing MK IMS (red) without (G) or with (H) L-plastin knockdown. Scale bar, 5 μm. (I) Quantification of MK IMS without or with L-plastin knockdown. IMS is filled with red color using binary editor function of Nikon NIS-Element software (n = 3). At least 15 MKs per condition per cord were analyzed.
L-plastin regulates MK migration, podosome formation, and IMS. (A) MK migration in transwells toward the SDF-1α chemoattractant on day 13. CD41a+CD42a+ MKs quantified by flow cytometry and normalized to no SDF-1α (n = 3). (B-C) Representative F-actin staining of spread MKs on day 11 by total internal reflection fluorescence (TIRF) microscopy for (B) control MKs and (C) MKs with L-plastin knockdown. (D) Quantification of MK podosomes per MK without or with L-plastin knockdown on day 11 by TIRF (n = 3). At least 25 MKs per condition per cord were analyzed. (E) Representative image of day 14 cultured MKs were stained for actin using ActinRed 555 ready probes (shown in red) and Wiskott-Aldrich syndrome protein (WASp) using anti-WASP antibody Alexa Fluor 488 (shown in green). Scale bar, 5 μm. (F) Representative image of day 14 cultured MKs on fibrinogen were stained for L-plastin using anti-L-plastin primary antibody and Alexa Fluor 555 seconday antibody (shown in red) and WASp stained with anti-WASP primary antibody and Alexa Fluor 488 seconday antibody (shown in green). White box shows magnified image. Scale bar, 10 μm. (G-H) Representative transmission electron microscopic images showing MK IMS (red) without (G) or with (H) L-plastin knockdown. Scale bar, 5 μm. (I) Quantification of MK IMS without or with L-plastin knockdown. IMS is filled with red color using binary editor function of Nikon NIS-Element software (n = 3). At least 15 MKs per condition per cord were analyzed.
We were struck by the appearance of actin aggregates in the MK where L-plastin had been knocked down (Figure 7B-C). These aggregates resembled podosomes, which are actin-rich structures important for MK interactions with matrix proteins and for penetration of PPs through the basement membrane.53 The increased numbers of MK podosomes after L-plastin knockdown was observed in MKs at both day 11 (Figure 7B-D) and day 14 of culture (supplemental Figure 11). The podosome marker, Wiskott-Aldrich syndrome protein,54 colocalized with actin (Figure 7E) and with L-plastin (Figure 7F). Lastly, because IMS formation depends on F-actin dynamics,22 we assessed the effect of L-plastin on the IMS area and observed significantly more invaginated membrane after L-plastin knockdown (Figure 7G-I; supplemental Figure 12 shows 36 additional images). Taken together, these data support that high levels of MK L-plastin in early MkPs enhance organized actin stress fibers and cell migration, and lower levels of L-plastin enhance MK IMS and promote podosome and PPF.
Discussion
Using an unbiased genome-wide transcriptomic approach, we identified MK miRs and mRNAs associated with MK differentiation and platelet number, and tested miR functionality in MKs derived from human HSPCs. The major findings in this study were that (1) miR-125a-5p is positively associated with human platelet number but not WBC or Hb levels, and enhances human MK PPF in vitro and murine platelet count in vivo; (2) miR-125a-5p directly targets and knocks down expression of the actin-bundling protein, L-plastin; (3) as MKs differentiate, miR-125a-5p and L-plastin levels increase and decrease, respectively; and (4) higher L-plastin levels in MkP promote migration, whereas decreased expression of L-plastin in mature MKs releases inhibition of MK IMS, and podosome and PPF. In addition, our work provides a public resource containing transcriptomes for primary MKs, platelets, and differentiating cultured MKs derived from human cord bloods. The actin cytoskeleton is acknowledged to play an important role in thrombopoiesis and platelet production, but often rather imprecise terms like “reorganization” describe this very complex process. Our findings add several novel molecular mechanisms and their interactions to the regulation of thrombopoiesis, and provide the first evidence for an F-actin–bundling protein as a regulator of MK PPF and thrombopoiesis.
Primary human MK, platelet, and CB MK transcriptome resource
The majority of platelet protein is synthesized in BM MKs, but the relationship between the peripheral blood platelet and BM MK transcriptomes is unknown. This is a highly relevant issue for understanding mechanisms accounting for platelet miR associations with physiologic traits. To address this specific issue, we profiled miR and mRNA from platelets and MKs from the same donors. Laser-captured microdissection of healthy donor BM aspirate slides enabled isolation of a pure population of mature MKs devoid of platelets that contaminate preparations generated by flow cytometry or density centrifugation. There were strong correlations between transcriptomes of primary BM MKs and platelets from the same donor, and between primary BM MKs and day 13 cultures of CD61+ MKs derived from TPO-stimulated cultures of cord blood HSPCs. Such associations enable generation of rational hypotheses for understanding novel molecular aspects of human physiology. Supplemental Tables 1-4 provide a resource for other investigators studying MK and platelet transcriptomes, including platelet miRs associated with platelet count in 154 healthy individuals and changes in transcriptomes as MKs proliferate and differentiate in response to TPO. Notably, these transcriptome findings may not apply to less mature MkPs.
miR-125a-5p, HSCs, MKs, and PPF
In vitro and in vivo overexpression and knockdown studies have identified miR-150, miR-155, miR-142-5p, or miR-146a as regulating megakaryocytopoiesis.55-59 None of these miRs have been correlated with human peripheral blood platelet number, and the role of MK miRs on late-stage human megakaryocytopoiesis and thrombopoiesis is poorly understood. An unbiased screen of 154 healthy donors suggested that miR-125a-5p may preferentially alter the MK lineage. The importance of PPs in platelet biogenesis is underscored by correlations between PP numbers and platelet counts in many inherited and chemokine- and drug-induced quantitative platelet disorders.60,61 Overexpression and knockdown of miR-125a-5p in human MKs demonstrated regulation of PPF in vitro. Consistent with this effect, in vivo inhibition of miR-125a-5p significantly lowered the platelet count in WT mice, although the effect was milder, perhaps due to species differences in miR-125a-5p or other off-target effects.
miR-125a-5p is part of an evolutionarily conserved cluster of 3 miR genes within 727 bp of one another on chr19q13.41, and all 3 miRs (miR-99b-5p, let-7e-5p, and miR-125a-5p) displayed increased expression during human MK maturation. However, compared with miR-125a-5p, inhibition of miR-99b-5p and let-7e-5p had less effect on MK PPF, and inhibition of all 3 simultaneously was no different than miR-125a-5p alone (supplemental Figure 13). Multiple murine mouse studies of miR-125a function have shown conflicting results on mouse platelet numbers. Sustained overexpression of the cluster or miR-125a alone has consistently shown increased numbers of long-term repopulating HSCs and the development of a myeloproliferative-like syndrome of leukocytosis, monocytosis, splenomegaly, and progressive anemia, with the majority of this overexpression effect being due to miR-125a-5p.46,47,49,62-64 None of these studies examined MKs, but Wojtowicz et al reported ∼35% higher platelet counts in mice overexpressing miR-125a-5p.49 Qin et al reported that a mir-125a−/− mouse had reduced granulopoiesis but no change in platelet counts, although a genomic characterization of the locus was not provided.65 Unlike most prior work, our study differs by its focus on human platelets and megakaryocytopoiesis, which may be relevant to the reported epidemiologic association between miR-125a-5p and ischemic stroke.66 Future mouse MK-specific knockout experiments may better clarify this issue.
Because miR-125a-5p levels decrease in differentiating mouse BM HSCs,46,47 we sought to determine the stage of MkP development in which miR-125a-5p might act to regulate platelet number. Using TPO-induced unilineage MK culture conditions, miR-125a-5p inhibition had no effect on the percentages of HSCs, HSPCs, or mature CD34−CD41+CD42+ MKs, but resulted in fewer day 13 MKs with PPs, suggesting that miR-125a-5p induces thrombopoiesis by acting on a later-stage MkP. The mechanism by which miR-125a-5p is upregulated during MK differentiation is unclear, but it is tempting to speculate that TPO enhances transcription through the flanking DNase I hypersensitivity sites (http://genome.ucsc.edu/ENCODE/releaseLog.html) and binding sites for STAT1, STAT5, or Ets family members known to regulate late MK differentiation.67-69
L-plastin regulates MK migration and PPF and branching
miR-125a-5p is predicted to target hundreds of MK genes, but only 3 emerged from our stringent filtering criteria. Although the miR-125a-5p effects on human thrombopoiesis may involve other MK genes, we were especially intrigued by the actin-bundling protein L-plastin as a potential mediator of MK PPF due to the importance of actin filament organization in platelet biogenesis.6,21 Plastins are a family of highly conserved actin-binding proteins that contain 2 predicted N-terminal calcium-binding domains and 2 C-terminal actin-binding domains, ABD1 and ABD2.70 L-plastin is a 65-kDa protein often reported as specific for leukocytes and some transformed cells. LCP1 expression is regulated by upstream repressor and steroid sex hormone receptors,71,72 but has not previously been shown to undergo miR regulation.
F-actin organization and dynamics regulate MK shape, migration, and early stage of PPF. We used gene editing of bulk MK cultures to obtain >95% knockdown of L-plastin and examined processes where actin has been linked to MK physiology and platelet production. Knockdown and overexpression of L-plastin increased and decreased, respectively, MK PPF, supporting its negative effect on this later stage of thrombopoiesis. Lower L-plastin levels induced greater PP branching, which is also consistent with the inverse association between platelet count and L-plastin levels. Perhaps greater F-actin bundling would increase PP stiffness and lower the PP bending required to form branches.
MkPs must migrate from the proliferative to the vascular niche to release platelets into the circulation. L-plastin is essential for lymphocyte polarization, lamellipodia formation, and motility toward chemokines CXLC12 and CXCL13.52,73 As MKs mature they migrate toward SDF-1 via its CXCR4 receptor, which increases day 6 to day 9 to day 13 (P = 10−12; supplemental Table 4). We showed that higher levels of L-plastin are associated with more migration. Cell migration involves stress fiber formation, which requires actin filament bundling. We found that normal MkP L-plastin levels contribute to MK stress fiber formation and migration. The apparently conflicting results between migration and PPF could be explained by the MkP need for thicker actin filament cables to achieve the strength required for high-tension stress fiber–mediated MK migration and to suppress premature release of platelets in the BM.74 At later stages of thrombopoiesis, such an L-plastin–mediated dense actin cytoskeleton could be inhibitory for invagination of plasma membrane to form the IMS or for protrusion of the IMS beyond the MK plasma membrane during initiation of PPF.
L-plastin regulates MK actin-dependent podosome and IMS formation
Actin filament organization regulates MK platelet production by its association with numerous structural and signaling molecules.6,21,24,75 Podosomes are observed at sites of cell contacts with the extracellular matrix and contain cores of F-actin that serve as a nidus for macromolecular-signaling clusters.76 Podosome assembly requires Rho GTPases, integrins, and actin-binding proteins, including L-plastin that binds β integrins via its C-terminal domain.77
Schachtner et al have shown that MK podosomes are required for MKs to extend protrusions across the basement membrane.53 We show that human MK L-plastin colocalizes with the podosome marker WASp. MK L-plastin knockdown induced more podosomes and a larger IMS, which provides the membrane for platelet biogenesis.15 These data support a model of MkP transition from an L-plastin–constrained migrating cell with less IMS and few podosomes to a spread, adherent mature MK with more podosomes giving rise to branching PPs. At least part of the reason podosomes form at late-stage megakaryocytopoieis and initiation of PPF may be the increased miR-125a-5p that lowers levels of L-plastin. Whether L-plastin directly or indirectly regulates podosome formation remains to be determined. It is possible, for example, that cessation of migration via L-plastin knockdown triggers other MK signals or gene expression to generate podosomes, which in turn proteolyze extracellular matrix allowing PPs to extend into the sinusoid.
In conclusion, the identification of miR-125a-5p as a regulator of platelet production may enhance in vitro manufacture of platelets.2,78,79 We have identified LCP1/L-plastin as a novel MK gene/protein regulating fundamental processes of cell migration and progeny formation. Proper organization of all cell actin cytoskeletons is critical to its normal and pathophysiologic function, and our work adds novel insight into this very complex process. These findings support future studies exploring the miR-125a-5p/L-plastin axis (1) in pathological conditions associated with thrombocytopenia and thrombocytosis and (2) as a potential therapeutic target for such disorders.
RNA-sequencing data are available in the Sequence Read Archive (SRA), accession number PRJNA642888.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Sara Hugo for technical assistance, Allie Grossman for histopathology analysis, Diana Lim for assistance with analysis of figures and software consultation, Linda Nikolova for technical assistance in the Electron Microscopy Core Laboratory, and Chris Rodesch and Michael J. Bridge of the Cell-Imaging Core, all at the University of Utah. The authors also thank Cory Lindsey and Xianguo Kong, and Lin Ma for technical assistance, at Thomas Jefferson University (Philadelphia, PA); the Pathology Core at Children’s Hospital of Philadelphia (CHOP; Philadelphia, PA) for technical assistance with laser-capture microdissections; and Renata Pellegrino Da Silva, Lifeng Tian, and Edward Liu from the Genomics Core, CHOP (Philadelphia, PA) for technical assistance with RNA sequencing.
This work was supported by National Institutes of Health, National Heart, Lung, and Blood Institute grant HL14124 and funding from the Division of Hematology and Hematologic Malignancies, University of Utah.
Authorship
Contribution: S.B., B.K.M., I.B., E.T., M.L.S., M.J.C., and R.A.C. performed experiments and analyzed the data; S.B. and P.F.B. designed research, interpreted results, and wrote the manuscript; L.C.E., S.C.M., S.N., A.S.W., J.W.R., R.M.O., M.T.R., R.A.C., and P.F.B. provided overall guidance; and all authors reviewed the manuscript
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Paul F. Bray, Division of Hematology and Hematologic Malignancies, Department of Internal Medicine, University of Utah, 15 North 2030 East, Eccles Institute of Human Genetics, Bldg 533 Room 4160B, Salt Lake City, UT 84112; e-mail: paul.bray@hsc.utah.edu.