Key Points
Secondary HLH is not due to primary cytotoxicity defects, but mimics other hyperinflammatory syndromes associated with a cytokine storm.

Abstract
Hemophagocytic lymphohistiocytosis (HLH) is a life-threatening hyperinflammatory condition. Primary HLH occurs early in life as a result of monogenic biallelic mutations affecting lymphocyte cytotoxicity. Secondary HLH occurs mostly in adults secondary to infection, lymphoma, or rheumatic disease. In this latter setting, lymphocyte cytotoxicity status is not known. We conducted a systematic evaluation of natural killer (NK) cell cytotoxicity in adult patients with secondary HLH. Adult patients with secondary HLH were prospectively studied ex vivo for total lymphocyte count and subtype, NK cell phenotype, perforin expression and degranulation, and natural or antibody-dependent cell cytotoxicity, in comparison with patients affected by the same underlying disease without HLH (disease controls [DCs]) and with healthy controls (HCs). Screening for variants of cytotoxity genes was systematically performed. 68 patients were included in the HLH group and 34 each in the DC and HC groups. In HLH patients, severe and transient lymphopenia, activated NK cell phenotype (eg, increased CD69, ICAM-1, HLADR, and CCR5 expression), and decreased capacity of interferon γ production were observed; mean perforin expression was normal; and degranulation tests and NK cell cytotoxicity were not different from those in DCs. A monoallelic variant of uncertain significance affecting a lymphocyte cytotoxicity gene or the perforin variant A91V was observed in almost 50% of the patients. We detected no major intrinsic cytotoxicity dysfunction in secondary HLH patients compared with DCs and no predicted pathogenic gene variant. The activated NK phenotype profile associated with decreased interferon γ production seems similar to those of other hyperinflammatory diseases such as sepsis or systemic juvenile idiopathic arthritis.
Introduction
Hemophagocytic lymphohistiocytosis (HLH) is a life-threatening syndrome characterized by a hyperinflammatory state associated with tissue lymphohistiocytic proliferation and hemophagocytosis.1 Diagnosis relies on the presence of several clinicobiological signs (eg, fever, organomegaly, cytopenia, hypertriglyceridemia, hyperferritinemia, and elevated serum soluble CD25 [sCD25] concentrations).2 Bone marrow hemophagocytosis is inconstant and nonspecific, but remains the hallmark of diagnosis.3
HLH can be divided into primary and secondary forms. Primary HLH usually occurs early in life as a result of recessive mutations of genes involved in T-lymphocyte and natural killer (NK) cell cytotoxicity.4 The best-known example is perforin deficiency characterizing familial HLH (F-HLH) type 2. Other mutations affecting various proteins involved in the secretion of perforin-containing granules may also induce the disease.4 Secondary or acquired HLH usually affects adults and may complicate various clinical situations, such as infection (eg, Epstein-Barr virus, cytomegalovirus, herpes simplex virus, varicella-zoster virus, and intracellular bacteria such as mycobacterial, fungal, or parasitic infection), malignancy (eg, non-Hodgkin T or B lymphoma), and rheumatic disease (mainly adult-onset Still’s disease [AOSD], systemic juvenile arthritis [sJIA], and systemic lupus erythematosus).5 In this rheumatologic setting, HLH is often referred to as macrophage activation syndrome.6
F-HLH pathogenesis has been studied using perforin-knockout animal models infected with lymphocytic choriomeningitis virus and is characterized by an overactive but partly ineffective immune system.7 Indeed, mutations affecting CD8 lymphocyte cytotoxicity impair the destruction of infected cells, leading to persistent antigenic stimulation, whereas the lack of NK cell cytotoxicity impairs the negative control exerted by these cells on CD8 lymphocyte and dendritic cell proliferation.8,9 These abnormalities induce an uncontrolled activation and expansion of interferon γ (IFN-γ)–producing CD8 lymphocytes, which subsequently induce macrophage activation, proliferation, and massive production of proinflammatory cytokines.10 Overproduction of IFN-γ and proinflammatory cytokines (ie, the cytokine storm) seems to characterize secondary HLH as well.11 However, the mechanisms underlying this hyperinflammatory state are not well known and may be variable, such as sustained Toll-like receptor activation by infectious or autoimmune triggers, as suggested by the cytosine guanine dinucleotide–activated Toll-like receptor 912 or the lipopolysaccharide-activated interleukin-6 (IL-6+/+) animal models,13 or may result from gain-of-function mutations affecting inflammasomes, such as the recently described NLRC4 inflammasome syndrome inducing IL-18 overproduction in humans.14,15 Although primary and secondary HLH seem to be 2 different entities with similar clinical phenotypes, some cases of F-HLH may be diagnosed in young adults,16 and many patients with secondary HLH may have compromised lymphocyte immune responses because of an underlying disease, such as HIV infection, or treatment with immunosuppressive drugs.17 Moreover, rare reports have suggested transient abnormal cytotoxicity in secondary HLH, sometimes associated with monoallelic variants of genes involved in F-HLH,18-20 as reported in patients with AOSD.21 However, no study has evaluated the real impact of these variants on the in vitro NK cell functions. We therefore conducted a prospective study of NK cell functions and genetic variants affecting CD8 and NK cell cytotoxicity in adult patients with secondary HLH.
Patients and methods
Patients
Patients with secondary HLH were prospectively included in this study between 2000 and 2014. We first enrolled patients with secondary HLH seen in the internal medicine department of the Conception University Hospital of Marseille between 2000 and 2004.22 The second inclusion period involved patients with secondary HLH hospitalized between 2010 and 2014 in several French hospitals (University Hospitals of Marseille and Nantes, Aix-en-Provence, and Toulon; Programme Hospitalier de Recherche Clinique #24-17). All patients were age >18 years, and the diagnosis of HLH was made using the criteria of the International HLH Society.2 No patient had received immunosuppressive treatment before inclusion. Two control groups were prospectively recruited. The disease control (DC) group included patients with infection, lymphoma at diagnosis, or autoimmune or inflammatory disease during disease flare-up (eg, systemic lupus erythematosus or AOSD), but without evidence of HLH. The healthy control (HC) group included free volunteers recruited at the Clinical Investigation Center of Marseille. Peripheral blood samples were all taken at the time of diagnosis (during the first 24 hours) and before initiation of any specific treatment. They were obtained after informed personal consent. This study was approved by the local ethics committee of Marseille University/Assistance Publique–Hôpitaux de Marseille.
Immunological study
All patients underwent an sCD25 (IL-2 receptor α-chain) assay (ELISA Kit; R&D Systems, Abingdon, United Kingdom). Lymphocyte phenotypes were performed on fresh whole-blood samples with the BD Multitest TBNK 6C reagent. NK cells were defined as CD3−CD56+ cells. Monoclonal antibodies from Becton Dickinson (Le Pont de Claix, France) were used: AmCyan or fluorescein isothiocyanate (FITC)–conjugated anti-CD3 (immunoglobulin G1 [IgG1], SK7), V450-conjugated anti-CD56 (IgG1, B159), PerCP-Cy5.5–conjugated anti-CD16 (IgG1, 3G8), APC-H7–conjugated anti-CD25 (IgG1, M-A251), phycoerythrin (PE)-Cy7–conjugated anti-CD69 (IgG1, FN50), PE-conjugated anti-bHLA-DR+ (IgG2a, L243), FITC-conjugated anti-CD94 (IgG1, HP-3D9), APC-conjugated anti-NKG2D (IgG1, 1D11), APC-conjugated anti-NKp46 (IgG1, 9E2), APC-conjugated anti-CXCR3 (IgG1, clone 1C6), AF647-conjugated anti-CCR2 (IgG2b, clone 48607), PC5-conjugated anti-CCR5 (IgG2a, clone 2D7), and PC7-conjugated anti-CCR7 (IgG2a, clone 3D12). PerCP-conjugated anti-NKG2C (IgG1, Fab 138C) was from R&D Systems. APC-conjugated anti-CD56 (IgG1, NKH-1), PE-conjugated anti-NKG2A (IgG2b, Z199), and PE-conjugated anti-NKp30 (IgG1, Z25) were from Beckman Coulter (Villepinte, France). Cells were analyzed by flow cytometry on a 3-laser FACSCanto II (BD) with FACSDiva software. Results were expressed as percentage of positive NK cells compared with specific corresponding isotype control among all NK cells or as median fluorescence intensity (MFI) of CD16, NKp30, or NKp46.
NK cell perforin expression
Whole blood was incubated for 30 minutes with CD45, CD3, and CD56. Red blood cells were lysed, fixed, and permeabilized according to the manufacturer’s recommendations. Cells were incubated at +4°C for 30 minutes with PE-conjugated antiperforin (IgG2b, clone DG9) or corresponding isotype control. After 2 washes, the cells were analyzed on a BD FACSCantoII cytometer. Results were expressed as percentage of perforin-positive NK cells or as MFI.
NK cell degranulation and NK cell IFN-γ production
The study was performed using peripheral blood mononuclear cells (PBMCs). We monitored NK cell activation by assessing the ability of these cells to degranulate and produce IFN-γ in response to several stimuli: major histocompatibility complex class I–negative human erythroleukemic K562 target cells (natural cytotoxicity) and mouse mastocytoma P815 cells coated with rabbit anti-mouse lymphocyte antibodies (Accurate Biochemicals, Westbury, NY; antibody-dependent cell cytotoxicity [ADCC]). Cell lines K562 and P815 were obtained from the American Type Culture Collection (Manassas, VA).
A total of 5 × 105 PBMCs per well were distributed in U-bottom 96-well plates and cultured in media alone or stimulated with target cells (2.5 × 105 per well) in the presence of Golgi Stop (1/1500; Becton Dickinson) and FITC-conjugated anti-CD107 monoclonal antibodies (anti-CD107a [IgG1, H4A3], anti-CD107b [IgG1, H4B4]; Becton Dickinson) for 4 hours. Cells were then washed in phosphate-buffered saline supplemented with 2% fetal calf serum and 1 mM of EDTA and stained for 30 minutes at 4°C with PerCP-Cy5.5–conjugated anti-CD3 antibody (BD), APC-conjugated anti-CD56 antibody (IgG1 N90; Beckman Coulter), and 2% normal mouse serum (Sigma Aldrich). After 2% paraformaldehyde fixation and permeabilization, IFN-γ production by activated NK cells was detected by incubation with PE-conjugated anti–IFN-γ antibody (IgG1, 4S-B3; BD) for 30 minutes at 4°C. For both CD107 and IFN-γ, results were expressed as the percentage of the whole NK cell population displaying positive staining.
NK cell cytotoxicity assay
To directly assess NK cell cytotoxicity, a flow cytometric assay based on staining with carboxyfluorescein diacetate succinimidyl ester (CFSE) target cells was used as previously described.23 Briefly, target cell suspensions (K562 and Ig-coated P815) were labeled with 0.5 µM of CFSE for 10 minutes at 37°C. After 2 washes, the CFSE-labeled target cells were resuspended in assay medium in a round-bottomed well plate. PBMCs were added at effector/target (E:T) ratios of 50:1, 25:1, 12:1, 6:1, and 3:1. Plates were incubated for 4 hours in a humidified atmosphere of 5% carbon dioxide at 37°C. Just before analysis, propidium iodide (2.5 µg/mL) was added to stain dead cells. Samples were directly analyzed by flow cytometry. Controls, including only CFSE-stained target cells, were assayed to determine spontaneous cell death. Dead target cells were identified as being double stained with CFSE and propidium iodide, and specific cytotoxicity was calculated as follows: (percentage of dead target cells at different E:T ratios − spontaneous dead target cells/100 − spontaneous dead target cells) × 100.
Genetic study
Targeted resequencing by next-generation sequencing (capture by hybridization approach) was performed on a panel of 9 HLH-associated genes (PRF1, UNC13D, STX11, STXBP2, SH2D1A, XIAP, RAB27A, LYST, and ITK). Illumina-compatible barecoded genomic DNA libraries were constructed according to the manufacturer’s sample preparation protocol (Ovation Ultralow; Nugen Technologies). Briefly, 1 to 3 µg of each patient’s genomic DNA was mechanically fragmented to a median size of 200 bp using focused ultrasonication; 100 ng of double-strand fragmented DNA was end repaired, and adaptors containing a specific 8-base barcode were ligated to the repaired ends (1 specific barcode per patient). DNA fragments were polymerase chain reaction amplified to obtain the final precapture barcoded libraries. The capture process was performed using homemade biotinylated probes, SureSelect reagents (Agilent), and 750 ng of the precapture barcoded library (or 750 ng of an equimolar pool of precapture libraries). The biotinylated single-strand DNA probes were designed and prepared to cover all the coding regions and intron-exon boundaries (>30 bases in the introns).
After demultiplexing, sequences were aligned to the reference human genome hg19 using the Burrows-Wheeler Aligner.24 For the 2 series of samples analyzed, the mean depth of coverage of the targeted exonic sequences per sample was ∼400× and 500× (copy number variant analysis of the exons is more accurate with a mean depth of coverage >250× to 300×), and 100% of the targeted bases were covered with >80 independent sequences (100% of the target at >80×).
Statistical analysis
Comparisons among the 3 groups were carried out using the nonparametric Kruskal-Wallis test for unpaired continuous data and Pearson χ2 test for categorical variables. The nonparametric Mann-Whitney U test was used to compare 2 groups for unpaired continuous data. Data were expressed as medians (10th-90th percentiles). A 2-tailed P value threshold of .05 was considered statistically significant.
An E:T ratio of 25:1 was selected to compare direct NK cell cytotoxicity. Because direct NK cell cytotoxicity is dependent on the number of NK cells among PBMCs, patient values for 3 levels of NK cell percentages (2%-5%, 5%-10%, and >10%) were compared with normal values. The correspondence was carried out in the “gmatch” algorithm implemented in SAS software. We used the Wilcoxon nonparametric test for statistical analysis.
Results
Patient characteristics
The HLH group included 68 patients, and the DC and HC groups included 34 patients each. Demographic, clinical, and biological data are listed in Table 1. HLH patients were mildly but not significantly older (median age, 58 years; min-max, 28-91) than DCs (median age, 56 years; min-max, 25-81) and HCs (median age, 51 years; min-max, 37-76). Underlying diseases in the HLH vs DC groups were comparable (including lymphoma subtypes), but the number of cases of AOSD flare-up was higher in the DC group (6 vs 3 in HLH group; P = .04). There was only 1 HLH related to Epstein-Barr virus infection.
Fever and organomegaly were statistically more common in HLH patients, as were cytopenia, hypertriglyceridemia, and hyperferritinemia (P < .001). Fibrinogen concentrations, although lower in the HLH group, were not significantly different from those in the DC group (P = .07), whereas sCD25 concentrations were higher in the HLH than in the DC group (P = .002). Interestingly, hemophagocytosis was present in 90% of HLH bone marrow aspirates, but in none of the 8 patients with lymphoma from the DC group who had undergone bone marrow aspiration.
Severe but transient NK lymphopenia is present in HLH patients
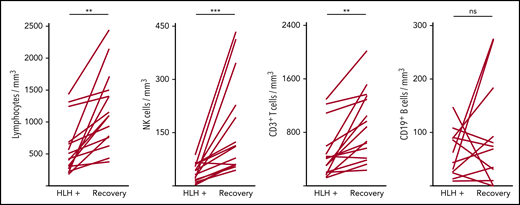
Severe lymphopenia was observed more frequently in the HLH group compared with the 2 control groups (Figure 1; supplemental Table 1A), affecting T, B, and NK cells. Among the 68 HLH patients, 15 had a lymphocyte phenotype after HLH recovery, showing that total lymphocyte, CD3, CD4, CD8, and NK cell counts returned to normal values after HLH cure (Figure 2; supplemental Table 1B). B lymphocyte profiles were more difficult to evaluate, because 4 HLH patients received rituximab during the course of their treatment.
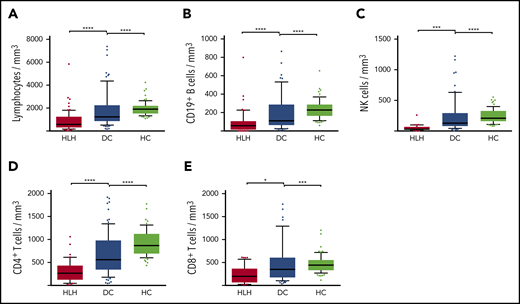
Lymphocyte counts in the 3 groups. Total lymphocytes (A), CD19+ B cells (B), NK cells (C), CD4+ T cells (D), and CD8+ T cells (E) in HC group (white plots), DC group (light gray plots), and HLH group (dark gray plots). Each point represents an outlier (error bars, 10th and 90th percentiles). *P < .05, ***P < .001, ****P < .0001 by Mann-Whitney U test.
Lymphocyte counts in the 3 groups. Total lymphocytes (A), CD19+ B cells (B), NK cells (C), CD4+ T cells (D), and CD8+ T cells (E) in HC group (white plots), DC group (light gray plots), and HLH group (dark gray plots). Each point represents an outlier (error bars, 10th and 90th percentiles). *P < .05, ***P < .001, ****P < .0001 by Mann-Whitney U test.
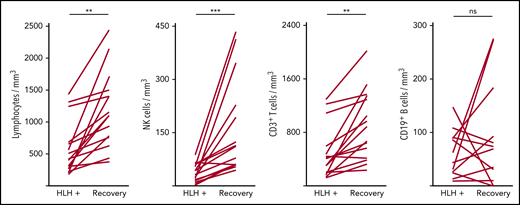
Evolution of lymphocyte counts during (HLH+) and after HLH recovery in 15 patients. Each line represents 1 patient. Lymphopenia (A) and NK (B) and T-cell (C) deficiency were corrected, but not B cells (D). **P < .01, ***P < .001 by Mann-Whitney U test compared with respective controls. ns, not significant.
Evolution of lymphocyte counts during (HLH+) and after HLH recovery in 15 patients. Each line represents 1 patient. Lymphopenia (A) and NK (B) and T-cell (C) deficiency were corrected, but not B cells (D). **P < .01, ***P < .001 by Mann-Whitney U test compared with respective controls. ns, not significant.
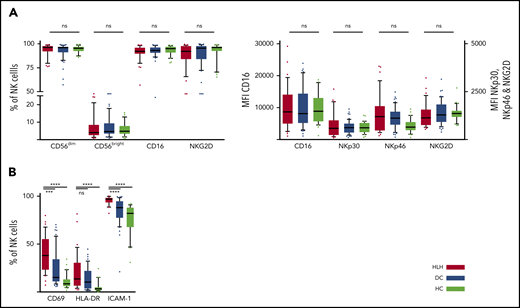
We then concentrated on NK cells and performed detailed phenotyping during the HLH episode. Distinct NK cell subsets have been described according to the expression of CD56; the CD56bright subset is less mature than the CD56dim subset, which is more efficient. CD56dim and CD56bright subsets were comparable in the 3 groups. No differences were observed in natural cytotoxicity receptors (NKp30 and NKp46) or in NKG2 receptors or CD16, the main receptor for ADCC (Figure 3A; supplemental Figure 1). Activation markers such as CD69, HLA-DR, and ICAM-1 were overexpressed on NK cells during HLH (Figure 3B). Similarly, the CCR5 homing receptor was overexpressed on NK cells from HLH patients, whereas no difference was observed between the 3 groups regarding the expression of others chemokine receptors (supplemental Figure 1). Similar activation markers such as CD69 and HLA-DR were also overexpressed on T cells during HLH (supplemental Figure 2).
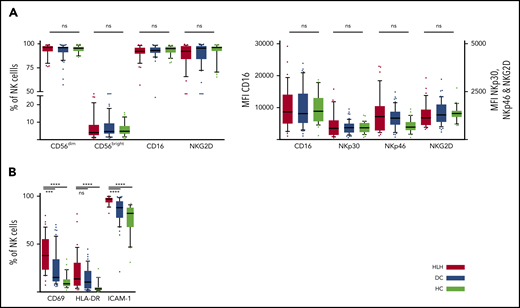
NK cell phenotyping in the 3 groups. Each point represents an outlier (error bars, 10th and 90th percentiles). ***P < .001, ****P < .0001 by Mann-Whitney U test (2 groups) and Kruskal-Wallis test (3 groups). ns, not significant.
NK cell phenotyping in the 3 groups. Each point represents an outlier (error bars, 10th and 90th percentiles). ***P < .001, ****P < .0001 by Mann-Whitney U test (2 groups) and Kruskal-Wallis test (3 groups). ns, not significant.
NK cell perforin expression is normal in most HLH patients
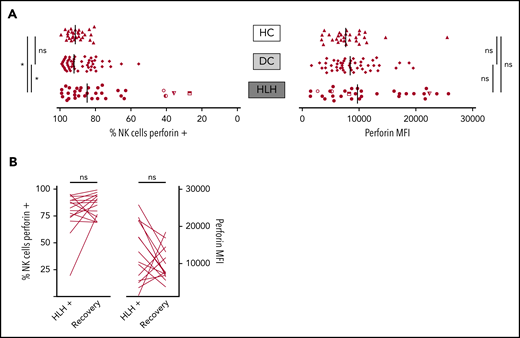
When considering perforin MFI, no significant difference was observed between HLH and control groups (median, 9733; 10th-90th, 3037-23 184 in HLH vs median, 8436; 10th-90th, 4127-16 523 in DC and 7664; 10th-90th, 3730-11 343 in HC; P = .233). However, the HLH patient group displayed fewer NK cells expressing perforin than the 2 control groups (median, 84.7%; 10th-90th, 40.8%-97.8% in HLH vs median, 92.1%; 10th-90th, 76.1%-98.1% in DC and 91.4%; 10th-90th, 82.5%-96.4% in HC; P = .033; Figure 4A). This minor difference was explained by the fact that in 4 HLH patients, <50% of their NK cells expressed perforin. Of note, 1 of these patients could be tested during and after recovery from the HLH episode. The percentage of NK cells expressing perforin was restored after HLH cure (Figure 4B).
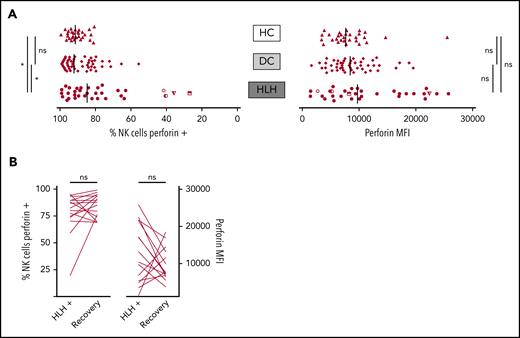
NK cell perforin expression in the 3 groups. (A) Percentage of NK cells expressing perforin (left) and perforin MFI (right). Each point represents 1 patient; bars indicate median values. (B) Evolution of the percentage of NK cells expressing perforin (left) and perforin MFI (right) during (HLH+) and after HLH recovery in 15 and 14 patients, respectively. Each line represents 1 patient. *P < .05 by Mann-Whitney U test (2 groups) and Kruskal-Wallis test (3 groups). ns, not significant.
NK cell perforin expression in the 3 groups. (A) Percentage of NK cells expressing perforin (left) and perforin MFI (right). Each point represents 1 patient; bars indicate median values. (B) Evolution of the percentage of NK cells expressing perforin (left) and perforin MFI (right) during (HLH+) and after HLH recovery in 15 and 14 patients, respectively. Each line represents 1 patient. *P < .05 by Mann-Whitney U test (2 groups) and Kruskal-Wallis test (3 groups). ns, not significant.
NK cell degranulation is normal in HLH patients
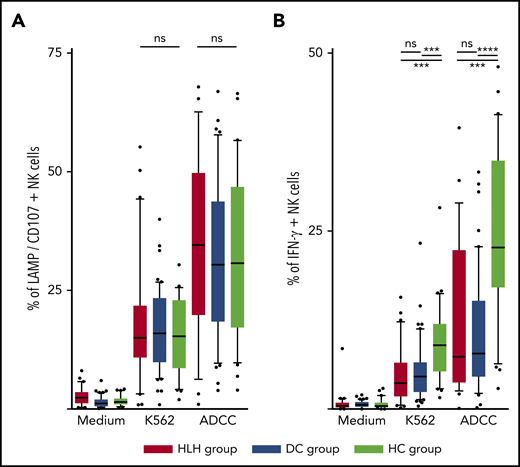
The ability of NK cells to degranulate was equivalent between HLH and the 2 control groups, against both K562 targets (median, 15%; 10th-90th, 3.2%-44.2 in HLH vs median, 16%; 10th-90th, 6.4%-26.8% in DC and 15.3%; 10th-90th, 4.1%-26.6% in HC; P = .8) and Ig-coated P815 targets (median, 34.6%; 10th-90th, 6.3%-62.6% in HLH vs median, 30.4%; 10th-90th, 9.7%-57.8% in DC and 30.8%; 10th-90th, 9.8%-56.6% in HC; P = .73; Figure 5A).
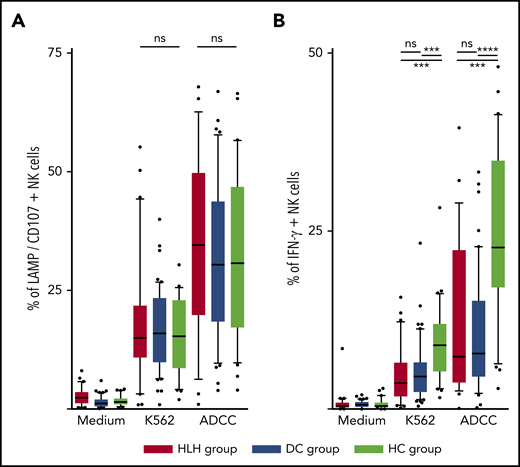
NK cell functional tests. (A) Degranulation evaluated by CD107/LAMP expression on NK cell surface after activation by K562 and Ig-coated P815 target cells. (B) IFN-γ–producing NK cells after activation by K562 and Ig-coated P815 target cells. Each point represents an outlier (error bars, 10th and 90th percentiles; box plots, median values and 25th and 75th percentiles). ***P < .001, ****P < .0001 by Mann-Whitney U test (2 groups) and Kruskal-Wallis test (3 groups). ns, not significant.
NK cell functional tests. (A) Degranulation evaluated by CD107/LAMP expression on NK cell surface after activation by K562 and Ig-coated P815 target cells. (B) IFN-γ–producing NK cells after activation by K562 and Ig-coated P815 target cells. Each point represents an outlier (error bars, 10th and 90th percentiles; box plots, median values and 25th and 75th percentiles). ***P < .001, ****P < .0001 by Mann-Whitney U test (2 groups) and Kruskal-Wallis test (3 groups). ns, not significant.
However, compared with those from HCs, NK cells from HLH patients had a weaker capacity to produce IFN-γ in response to activation with K562 and Ig-coated P815 targets (P < .001), but there was no difference between HLH patients and DCs, suggesting that this phenomenon was probably related to the underlying disease rather than to HLH itself (Figure 5B).
NK cell cytotoxicity is normal in HLH patients
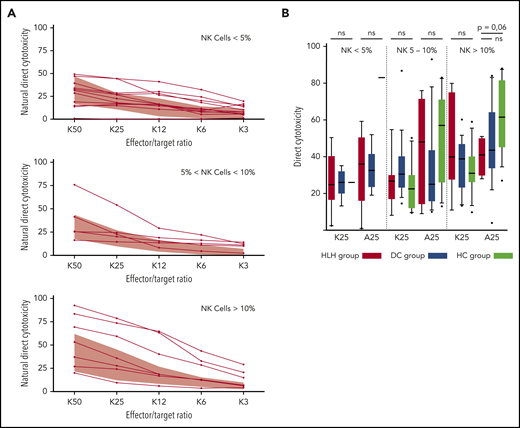
Because some patients had <2% NK cells, direct NK cell cytotoxicity could only be studied in 25 HLH patients and compared with 30 patients from the DC or HC group. In addition, matching with the NK cell count was absolutely necessary to differentiate between a cytotoxic alteration related to a low percentage of NK cells (quantitative abnormality) vs an inability of NK cells to lyse their targets (functional abnormality).
HLH patients showed no deficiency of cytotoxicity when studying natural cytotoxicity (statistical analysis based on E:T ratio of 25:1 [K25]) and ADCC (statistical analysis based on same E:T ratio [A25]; Figure 6A). After matching on the percentage of NK cells present in the total lymphocyte population, NK cell cytotoxicity seemed to be equivalent in the 2 different groups of patients and HCs (Figure 6B). Of note, a unique patient showed a severe NK cell cytotoxicity deficiency. This patient was a 91-year-old woman with severe and idiopathic HLH who died as a result of the disease and carried a monoallelic polymorphism of PRF1 (Table 2; patient 11).
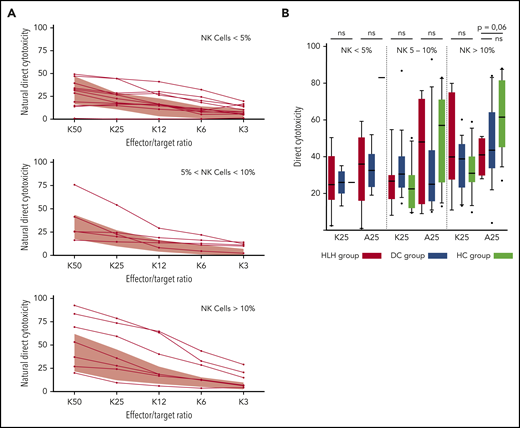
Direct NK cell cytotoxicity depending on cell percentage (<5%, 5%-10%, >10%) of total lymphocytes. (A) Natural cytotoxicity is represented for the different E:T ratios. Each line represents 1 HLH patient. The gray waves represent normal values derived from HCs. (B) Evaluated in an E:T ratio of 25:1 (K25 for K562 targets and A25 for ADCC); no differences between the 3 groups. Each symbol represents an outlier (error bars, 10th and 90th percentiles). ns, not significant (P > .05) by Wilcoxon nonparametric test.
Direct NK cell cytotoxicity depending on cell percentage (<5%, 5%-10%, >10%) of total lymphocytes. (A) Natural cytotoxicity is represented for the different E:T ratios. Each line represents 1 HLH patient. The gray waves represent normal values derived from HCs. (B) Evaluated in an E:T ratio of 25:1 (K25 for K562 targets and A25 for ADCC); no differences between the 3 groups. Each symbol represents an outlier (error bars, 10th and 90th percentiles). ns, not significant (P > .05) by Wilcoxon nonparametric test.
Genetic study
DNA-targeted resequencing of the 9 HLH genes was performed in 28 HLH patients. For all the patients, coverage of each gene was >30×. Overall, 582 unique variants were identified in the 28 patients. After filtering to exclude synonymous, intronic, and untranslated region variations, 42 variants remained. Filters were then applied to preserved variants with a possible impact at the protein level or with a minor allele frequency (<5%) in the Exome Aggregation Consortium data set (https://gnomad.broadinstitute.org; supplemental Table 2A). After filtering, 26 variants remained in 4 different genes (LYST, PRF1, STX11, and UNC13D), all monoallelic and distributed in a total of 15 patients. A majority of variants were found in LYST and UNC13D, likely reflecting gene size. No copy number variations were identified in the 9 HLH-related genes. Details of the variations are listed in Table 2 and supplemental Table 2B. A majority of the patients (n = 11 of 15) carried only 1 variant. Three patients carried a variant in 3 different genes, with only 1 variant predicted to be damaging by either SIFT or PolyPhen-2. One patient carried 7 monoallelic variants, all in the LYST gene, none of them with an in silico damaging prediction. Among the 5 possibly damaging variants identified in the patient cohort, the variant PRF1 c.272C>T p.Ala91Val was found in 2 patients in a heterozygous state, with a frequency of 3.6%. This allele frequency does not significantly differ from that reported in the European population (4.6%). The 91-year-old patient with severe HLH who had abnormal cytotoxicity tests was found to have a monoallelic benign variant of PRF1 (c.A755G; p.N252S; supplemental Table 2; patient 11). We then looked for the potential impact of these different variants on NK cell perforin expression and degranulation activity or on disease severity. As shown in Table 2, we did not find any correlation between the number or putative severity of the variants identified in the patients or the functional parameters. Similarly, no correlation was observed with disease severity, and among the 4 patients who died during the course of the study, no variant was identified in 2 of them.
Discussion
This study prospectively evaluated NK phenotype and function during secondary HLH. Compared with HCs or to patients with similar underlying disease, we observed transient modifications such as a global lymphopenia, decreased NK cell count with an activated phenotype, and decreased capacity of NK cells to secrete IFN-γ. However, NK cell degranulation and cytotoxic functions seemed to be normal overall. In agreement, sequencing of the various F-HLH genes revealed no biallelic mutations. However, a monoallelic variant affecting 1 or several F-HLH genes was found in almost 50% of the patients, despite normal in vitro functional tests. Among the 5 possibly damaging variants identified in the patient cohort, PRF1 c.272C>T p.Ala91Val was found in 2 patients in a heterozygous state. The A91V PRF1 variant is present in 3% to 18% of the European population,25,26 rendering our genetic results somewhat expectable. Although this variant has previously been reported to partially impair perforin stability27,28 and NK cell cytotoxicity,29 our patients had no cytotoxicity defects in vitro. This observation may seem contradictory to several published retrospective series of patients age >13 years reporting up to 15% of monoallelic variants, half of them involving the perforin gene, in which in vitro cytotoxicity was sometimes performed and found to be abnormal.20 However, these previous results were surprising, because heterozygous parents of children with F-HLH usually never develop the disease. It should also be noted that these series were based on a retrospective registry, and no details were provided concerning the in vitro functional tests performed, notably whether cytotoxicity was studied after normalization of NK cell count. This seems to be a critical point, and a similar observation has been made by others regarding HLH-complicating systemic JIA.30 Overall, our results suggest that either monoallelic variants have no critical impact on cytotoxicity or the commonly used in vitro cytotoxicity tests may not be accurate enough to detect milder impairment.
We should remain cautious, however, before concluding that monoallelic variants do not play any role in secondary HLH pathogenesis. First, the number of our patients was too small to detect any correlation. Moreover, data obtained in F-HLH animal models argue in favor of polygenic disease. Accumulation of variants in mice has been shown to impair NK cell cytotoxicity in vivo, increasing the risk of HLH.31 In humans, biallelic hypomorphic variants affecting cytotoxicity have been reported to be associated with late-onset HLH, despite the fact that in vitro cytotoxicity is not fully abolished.32 Similarly, in patients with sJIA, those who developed secondary HLH carried more monoallelic variants associated with F-HLH.21,33 In addition, in vitro tests may not be representative of the situation in vivo. Some heterozygous perforin variants have been shown to decrease protein stability and function at high temperature,34 possibly in inflammatory conditions. Moreover, we only screened genes known to be involved in cytotoxicity and F-HLH. A pathogenic intronic noncoding variant affecting UNC13D has been reported to be responsible for F-HLH type 3.35 A recent report found that almost 20% of young patients with HLH have no mutations in genes involved in lymphocyte cytotoxicity, but rather variants in genes involved in the innate immune response, such as NLRC4, NLRP12, NLRP4, NLRC3, and NLRP13.36 HLH may not be limited to impaired cytotoxicity, but may also involve the capacity to develop a more severe inflammatory response.
Other lymphocyte abnormalities were observed in secondary HLH patients. A circulating transient lymphopenia, notably affecting NK cells, was an observation made in this study that seems consistent with animal models of secondary HLH.37-39 NK cells in our patients expressed significantly higher surface activation markers, such as CD69, HLADR, or ICAM-1, but overall their phenotype seemed normal, and no NK subpopulation was significantly modified. Four of our patients had a decreased number of NK cells expressing perforin, which recovered after HLH resolution in the only patient in whom it could be measured. These data seem quite similar to previous observations made in patients with sJIA/AOSD. In this disease, secondary HLH affects 10% of patients,40,41 and several NK disorders have been reported: NK lymphopenia, modified NK phenotype, decreased perforin expression, and decreased cytotoxicity.28,42-45 Decreased cytotoxicity, however, seems questionable, because in the only study in which a normalization of NK number was seen, no significant cytotoxicity decrease was observed.30 sJIA/AOSD and secondary HLH are both characterized by a hyperinflammatory state with high serum concentrations of inflammatory cytokines, notably IL-1830,44 and IL-6.37,43 IL-18 is known to activate NK cell functions, but IL-18 receptor signaling seems to be impaired in sJIA/AOSD, possibly explaining NK lymphopenia and moderate dysfunction. NK lymphopenia has also been observed in a rare genetic form of HLH resulting from NLRC4 inflammasome mutations, characterized by very high concentrations of circulating IL-18.46 Similarly, human NK cells exposed to high concentrations of IL-6 were shown to have impaired cytotoxicity with reduced perforin expression.38 Overall, these data in secondary HLH are reminiscent of observations made in various hyperinflammatory states, such as sepsis, systemic inflammatory response syndrome, or fatal H1N1 virus infection.47,48 In sepsis, notably, the cytokine storm is compensated for by a reprogramming response called tolerization,49 characterized by transient global lymphopenia, decreased NK cell number, and ability to produce IFN-γ,50 as observed in our patients.
In conclusion, our study suggests that secondary HLH in adults is different from F-HLH and is not a monogenic disease associated with loss of lymphocyte cytotoxicity. Monoallelic variants were observed in 50% of patients, but some are common in the general population and did not affect degranulation or cytotoxicity functions tested in vitro. Therefore, testing of perforin expression and degranulation remain pivotal investigations in young adults affected by HLH, but with increasing age, the chances of discovering cytotoxic abnormalities using these tests decrease. Patients with secondary HLH seem to have transient NK cell alterations, which may be common to other hyperinflammatory states, such as sepsis or systemic inflammatory response syndrome.
For an access to individual participant data, please contact julien.carvelli@ap-hm.fr.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
This work was supported by grants from the Programme Hospitalier de Recherche Clinique National in 2007 (#24-17) and from Agence Nationale de la Recherche in 2007 (#MRAR-019-02).
Authorship
Contribution: G.K. devised and supervised the study and took care of several patients; J.C., C.P., C.F., F.V., and G.K. performed research with the help of the coauthors; J.C. and G.K. wrote the manuscript with the help of the other coauthors; C.P., C.F., S.A., P.N., C.B.-F., and G.d.S.-B. performed experiments; J.C., C.P., M.B., G.d.S.-B., and G.K. analyzed the data; and F.V., K.M., A.C., M.H., J.-R.H., G.d.S.B., and the HLH-2007 Study Group provided key expertise and samples.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
A list of members of the HLH-2007 Study Group appears in “Appendix.”
Correspondence: Gilles Kaplanski, Hopital de la Conception, 147 boulevard Baille, Marseille, 13005 France; e-mail: gilles.kaplanski@ap-hm.fr; and Julien Carvelli, Hopital de la Conception, 147 boulevard Baille, Marseille, 13005 France; e-mail: julien.carvelli@ap-hm.fr.
Appendix
The members of the HLH-2007 Study Group are: Régis Costello (Service d’Hématologie et Thérapie Cellulaire, Hôpital de la Conception, AP-HM, Marseille, France); Nathalie Costedoat-Chalumeau (Service de Médecine Interne, Hôpital Cochin, Assistance Publique–Hôpitaux de Paris, Paris, France); Hacene Fezoui (Service d’Hématologie, Centre Hospitalier [CH] de Toulon, Toulon, France); Jean-Pierre De Jaureguiberry, Philippe Carli, Gilles Defuentes, and Jean-François Paris (Service de Médecine Interne, Hôpital d’Instruction des Armées Sainte-Anne, Toulon, France); Thierry Allegre (Service d’Hématologie et Immunologie, CH du Pays d’Aix, Aix-en-Provence, France); Régis Kaphan (Service de Médecine Interne et Oncologie, CH de Cannes, Cannes, France); Yael Berda-Haddad (Service d’Hématologie, Hôpital de la Conception, AP-HM, Marseille, France); and Sophie Morange (Centre d’Investigation Clinique, Hôpital de la Conception, Marseille, France).