Abstract
Warm autoimmune hemolytic anemia (wAIHA) is caused by increased erythrocyte destruction by immunoglobulin G (IgG) autoantibodies, with or without complement activation. Antibody-dependent cell-mediated cytotoxicity by macrophages/activated lymphocytes occurs in the lymphoid organs and spleen (extravascular hemolysis). The ability of the bone marrow (BM) to compensate determines clinical severity. The different pathogenic mechanisms, their complex interplay, and changes over time may explain wAIHA’s great clinical heterogeneity and unpredictable course. The disease may be primary, drug induced, or associated with lymphoproliferative neoplasms, autoimmune and infectious diseases, immunodeficiencies, solid tumors, or transplants. Therapeutic interventions include steroids, splenectomy, immunosuppressants, and rituximab; the latter is increasingly used in steroid-refractory cases based on evidence from the literature and a few prospective trials. We present 5 patient case studies highlighting important issues: (1) the diagnosis and proper use of steroid therapy, (2) the concerns about the choice between rituximab and splenectomy in second-line treatment, (3) the need of periodical re-evaluation of the disease to assess the possible evolution of relapsed/refractory cases in myelodysplastic and BM failure syndromes, and (4) the difficulties in managing cases of severe/acute disease that are at high risk of relapse. Incorporating novel targeted therapies into clinical practice will be an exciting challenge in the future.
Introduction
Warm autoimmune hemolytic anemia (wAIHA) is the most prevalent form of autoimmune hemolytic anemia (AIHA), accounting for 60% to 70% of all cases. It is usually due to an immunoglobulin G (IgG) autoantibody that may activate complement (C) if present at high titer or if IgG1 and IgG3 subclasses are prevalent. The cornerstone of diagnosis is the direct antiglobulin test (DAT), which may be positive with anti-IgG antisera (70% of all wAIHA) or anti-IgG plus C at low titer. Antibody-dependent cell-mediated cytotoxicity (ADCC) is the main driver of red blood cell (RBC) destruction, occurring in the lymphoid organs and spleen. Additional pathophysiologic mechanisms include autoreactive cellular effectors (T cells, macrophages), unbalanced CD4+ regulatory T cells and cytokines, and, most importantly, the ability of the bone marrow (BM) to compensate. All of these factors, as well as their complex interplay and changes over time, may explain the great clinical heterogeneity and unpredictable course of AIHA.1-3
Moreover, wAIHA may be primary, or associated with a variety of conditions, such as lymphoproliferative neoplasms, autoimmune and infectious diseases, immunodeficiencies, solid tumors, or transplants, or with drugs, including the novel checkpoint inhibitors.3,4 Therapies used to treat wAIHA are based on limited evidence derived from expert opinion5-9 and a small number of prospective trials.10-12 Over the last 2 decades, steroids, splenectomy, and immunosuppressants have been the mainstays of treatment. More recently, rituximab has increasingly been used, and experimental therapies directed at complement, proteasomes, and various kinases are under development.9,13 Below, we present 5 patient case studies that highlight important issues regarding diagnosis and first-line steroid therapy, the choice of second-line treatment, the potential for relapsed or refractory (R/R) cases of AIHA to evolve into myelodysplastic syndromes (MDSs) and BM failure (BMF), and difficulties in the management of severe or acute wAIHA.
The pitfalls of diagnosis and first-line steroid therapy
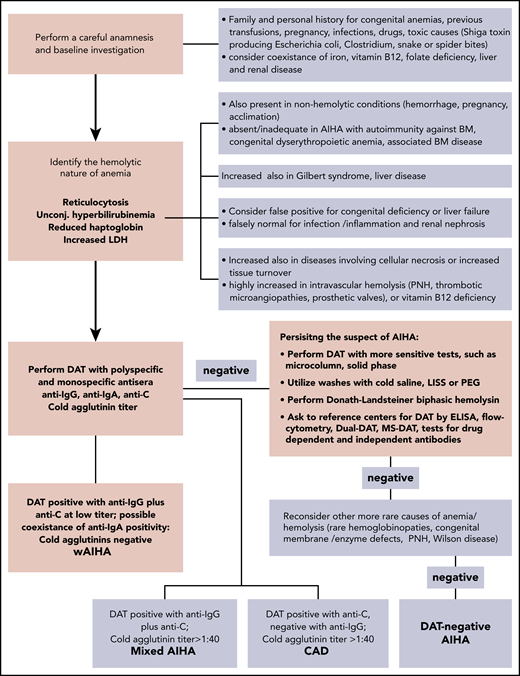
The DAT is the cornerstone of AIHA diagnosis. However, 5% to 10% of patients with clear evidence of the disease may test negative, despite extensive evaluation with methods more sensitive than the standard tube (microcolumn, solid phase, washings with cold or low-ionic salt solutions) (Figure 1). Moreover, it may be falsely positive in many healthy subjects, after some therapies (IV immunoglobulins, Rh immune globulins, antithymocyte globulins, daratumumab), in conditions with paraproteins or elevated serum globulins, or in recently transfused patients (alloantibodies), and may also remain positive in patients with AIHA in remission.3,14,15 Thus, AIHA cannot be diagnosed from DAT positivity alone; there should be evidence of hemolysis, and other congenital or acquired hemolytic disorders should be excluded in complex cases. Furthermore, confounding factors should always be considered (Figure 1).
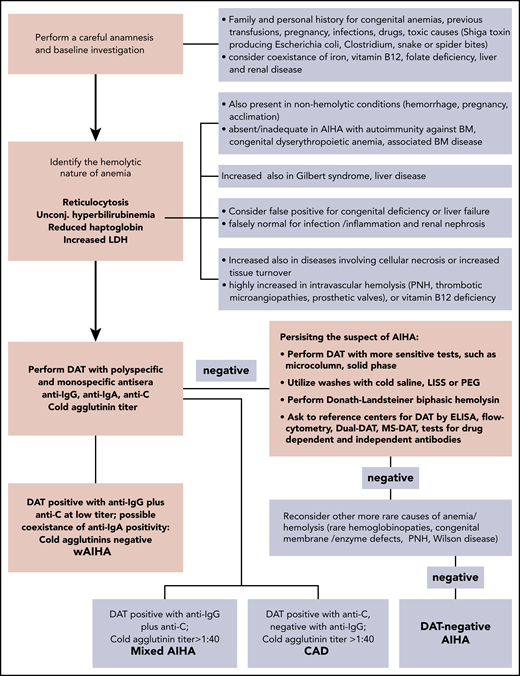
Diagnostic algorithm of AIHA. The DAT or Coombs test is the cornerstone of diagnosis, and allows the distinction of the different forms of AIHA. wAIHA is the most common form, accounting for 60% to 70% of all cases; the DAT is positive with anti-IgG antisera (70% of all wAIHA) or anti-IgG plus C at low titer. Cold agglutinin disease (CAD; 20% to 25% of all AIHAs) is characterized by DAT positivity with anti-C antisera and high titer of cold agglutinins. In mixed forms (5% to 10% of all AIHAs), the DAT is positive for IgG plus C, and cold agglutinins are present at high titer. The atypical forms (∼10% of all AIHAs) include DAT−, IgA, and warm IgM-driven AIHAs. Finally, it is necessary to record the very rare form named paroxysmal cold hemoglobinuria (1% to 3% of all AIHAs) sustained by the biphasic Donath-Landsteiner hemolysin. ELISA, enzyme-linked immunosorbent assay; LISS, low-ionic salt solution; MS-DAT, mitogen-stimulated DAT; PEG, polyethylene glycol; PNH, paroxysmal nocturnal hemoglobinuria.
Diagnostic algorithm of AIHA. The DAT or Coombs test is the cornerstone of diagnosis, and allows the distinction of the different forms of AIHA. wAIHA is the most common form, accounting for 60% to 70% of all cases; the DAT is positive with anti-IgG antisera (70% of all wAIHA) or anti-IgG plus C at low titer. Cold agglutinin disease (CAD; 20% to 25% of all AIHAs) is characterized by DAT positivity with anti-C antisera and high titer of cold agglutinins. In mixed forms (5% to 10% of all AIHAs), the DAT is positive for IgG plus C, and cold agglutinins are present at high titer. The atypical forms (∼10% of all AIHAs) include DAT−, IgA, and warm IgM-driven AIHAs. Finally, it is necessary to record the very rare form named paroxysmal cold hemoglobinuria (1% to 3% of all AIHAs) sustained by the biphasic Donath-Landsteiner hemolysin. ELISA, enzyme-linked immunosorbent assay; LISS, low-ionic salt solution; MS-DAT, mitogen-stimulated DAT; PEG, polyethylene glycol; PNH, paroxysmal nocturnal hemoglobinuria.
Patient 1
A 49-year-old woman was referred to our center with presumed DAT− AIHA, present for 2 years, and diagnosed on the basis of her response to steroids and exclusion of other hemolytic conditions. Initially, hemoglobin (Hb) values were around 8 g/dL, and normalized after the first course of oral prednisone (1 mg/kg per day for 2 weeks, followed by quick tapering and discontinuation within 2 months). A few months after discontinuation, a first relapse occurred, again responsive to oral steroids, but requiring higher doses and longer administration (1.5 mg/kg per day for 4 weeks, followed by tapering and discontinuation within 4 months). Three months before referral, a third relapse occurred (Hb, 7.7 g/dL), again treated with steroids. DAT results were persistently negative. Upon referral, the patient was still on a high dose of steroids (0.7 mg/kg per day), with a partial response (Hb, 9.6 g/dL). Generalized Cushing habitus was present, with mild hyperglycemia and osteoporosis. The patient sought advice on a second-line therapy.
Commentary on patient 1
It is well established that ∼80% to 90% of wAIHA patients respond to a 3- to 4-week course of prednisone 1 to 2 mg/kg per day (Table 1). In steroid-responsive patients, the taper can begin after 14 to 21 days, keeping in mind that relapse is more common if corticosteroids are tapered to ≤10 mg in <2 months and stopped in <6 months.9,14,16 Thus, it is important not only to consider Hb values, but also to strictly monitor all hemolytic markers, and delay tapering if hemolysis is ongoing (Figure 2).
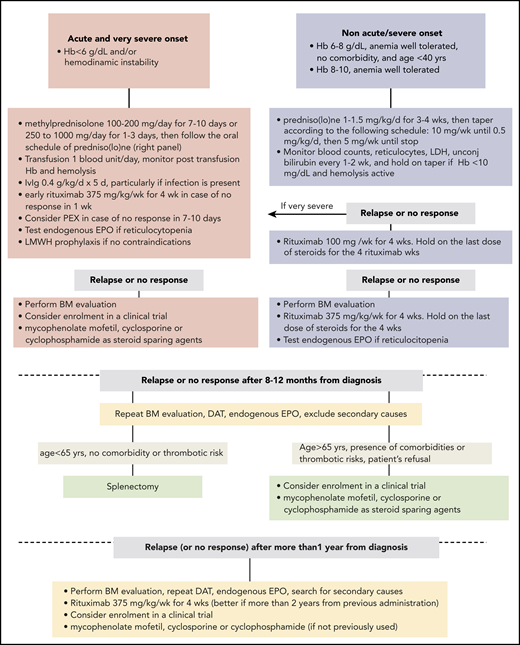
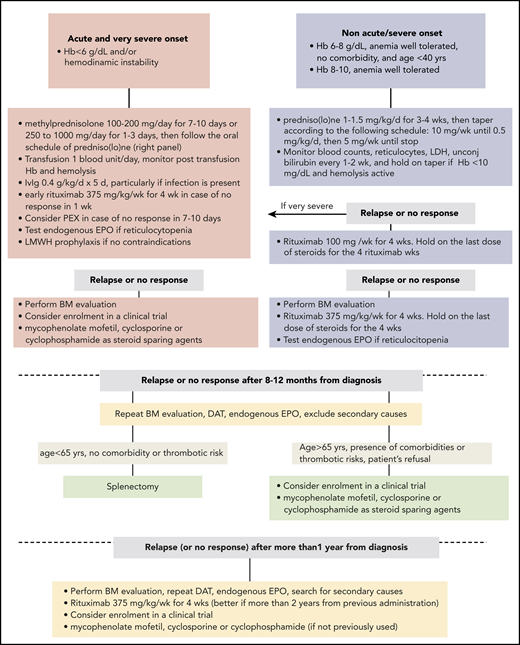
Therapeutic flow-chart of wAIHA. This figure illustrates the therapy lines of wAIHA. Responses include complete responses (CR) and partial responses (PR); CR is defined by normalization of Hb and hemolytic markers (unconjugated bilirubin, LDH, haptoglobin, and reticulocytes); PR is defined by Hb >10 g/dL or at least an increase by >2 g/dL, with or without biochemical resolution of hemolysis; lack of response or relapse is defined as Hb <10 g/dL or at least 2 g/dL decrease with alteration of hemolytic markers. EPO, erythropoietin; LMWH, low-molecular-weight heparin; PEX, plasma exchange.
Therapeutic flow-chart of wAIHA. This figure illustrates the therapy lines of wAIHA. Responses include complete responses (CR) and partial responses (PR); CR is defined by normalization of Hb and hemolytic markers (unconjugated bilirubin, LDH, haptoglobin, and reticulocytes); PR is defined by Hb >10 g/dL or at least an increase by >2 g/dL, with or without biochemical resolution of hemolysis; lack of response or relapse is defined as Hb <10 g/dL or at least 2 g/dL decrease with alteration of hemolytic markers. EPO, erythropoietin; LMWH, low-molecular-weight heparin; PEX, plasma exchange.
On the other hand, excessive and prolonged steroid therapy (>15 months) may result in significant side effects, particularly osteoporosis, even at doses considered low by hematologists (prednisolone, ∼7.5 mg per day for ≥3 months).17 Patient 1 had not received the recommended preventive actions (bone-mineral density screening, 800 IU of vitamin D daily, 700-1200 mg of daily calcium). Moreover, a quick tapering of steroids may have played some role in the first and second relapses. However, it is inappropriate to maintain a patient on steroids without considering second-line treatment; this cannot be fully justified by the uncertain diagnosis and persistently negative DAT.
Rituximab or splenectomy: which second-line therapy for wAIHA?
Rituximab is becoming the preferred second-line therapy for wAIHA, with an 80% overall response rate, a median response time of 3 to 6 weeks (with a range of 2 to 16 weeks), and a sustained response rate of ∼60% at 3 years (Table 1).18 It has been thoroughly researched and has proven effective in primary and most secondary forms of wAIHA, and retreatment appears similarly successful. Various doses have been used (375 mg/m2 per week for 4 weeks, most commonly; 100 mg weekly for 4 weeks, mainly in nonsevere forms and in the elderly; 1 g on days 1 and 15, particularly in wAIHA associated with other autoimmune diseases), with quite similar efficacy (response rate ∼80%, relapse-free survival of ∼60% at 3 years), although direct comparisons have not been made.9,10,19
An interesting approach is to use rituximab in first-line therapy: in a prospective randomized trial, rituximab with prednisolone increased the rate and duration of the response compared with prednisolone alone.11 Furthermore, a randomized double-blind trial demonstrated the efficacy and safety of rituximab vs placebo among newly diagnosed patients on standard prednisone therapy.12
However, rituximab is not available worldwide, nor is it universally available from public health services. Moreover, its significant side effects must be considered, particularly immunodeficiency (late-onset neutropenia, hypogammaglobulinemia) and reactivation of underlying infections (hepatitis B [HB] virus [HBV], hepatitis C [HCV], HIV, tuberculosis). Various tests (HB surface antigen [HBsAg], anti-HBs, anti-HBc, anti-HCV, anti-HIV, and quantiFERON-TB) are advisable before steroid therapy, but mandatory before rituximab. Lamivudine prophylaxis for up to 18 months is recommended for anti-HBc and/or anti-HBs antibody-positive patients (if not vaccinated).
Splenectomy is as effective as rituximab, with somewhat longer-lasting remissions (Table 1).8,9,16,20-22 Use of splenectomy has gradually declined, however, to <10% of cases, mainly due to the increased infection risk, particularly in the first year. This risk is not eliminated by vaccination and antibiotic prophylaxis. Notably, vaccination may result in a suboptimal response after immunosuppression, and is therefore recommended before commencing rituximab, in line with national guidelines.1,23
Patient 2
A 39-year-old man with a 15-year history of wAIHA was referred to our clinic after his fifth relapse (Hb, 8.5 g/dL; lactate dehydrogenase [LDH], 1.3 upper limit of normality [ULN]; reticulocytes, 125 × 109/L; unconjugated bilirubin, 2.7 mg/dL; DAT, IgG+). He had been treated with steroids in various cycles and schedules, alone or in association with azathioprine, cyclophosphamide, or rituximab, and had repeatedly refused splenectomy. Upon referral, the patient sought an effective and definitive therapy. Poor compliance became apparent, precluding his enrollment in trials of new drugs. After extensive discussion, he agreed to undergo splenectomy, and prophylactic vaccination was performed. Laparoscopic splenectomy was converted to laparotomic, for surgical reasons, and was complicated by abdominal wall hematoma, but resulted in a complete hematologic response. Four months after surgery, we learned that the patient died of overwhelming sepsis in a local hospital.
Commentary on patient 2
The most feared complication of splenectomy is overwhelming sepsis due to encapsulated bacteria, with a risk of 3% to 5% and a mortality rate of up to 50%, even after the introduction of vaccinations. The role and efficacy of antibiotic prophylaxis remains unclear, and not all experts recommend this approach.1,23 Given the patient’s poor compliance, continuous antibiotic therapy (although proposed) was probably not taken. Therefore, the patient’s level of education and awareness about complications should always be strongly pursued.
One drawback of splenectomy is the lack of reliable predictors of outcome because its effectiveness has not been related to disease duration, response to steroids, or the extent of splenic sequestration. Splenectomy may be preferred in younger patients, or in those who wish to become pregnant. Conversely, the following factors make splenectomy inadvisable: age (>65-70 years), cardiopulmonary disorder, previous history or serious risk of thrombosis, hepatitis C, underlying immunodeficiency, and lymphoproliferative and systemic autoimmune conditions. Moreover, splenectomy may be associated with surgical complications (pulmonary embolism, intra-abdominal bleeding, abdominal abscess, abdominal wall hematoma), although the laparoscopic approach has reduced the risk compared with open surgery.20,22
Unlike immune thrombocytopenia (ITP), in AIHA there is no consensus on the timing of splenectomy, even if clinical practice suggests a “neither too early nor too late” approach. In patient 2, long-standing immunosuppression may have hampered immunocompetence, predisposing him to harmful infections. However, no reliable markers of immunocompetence are available, and even after vaccination, protective antibody titers are difficult to establish. As the number of patients subjected to different types of immunosuppression therapy for various conditions increases, large prospective studies to address this knowledge gap are warranted. Historical immunosuppressants (azathioprine, cyclophosphamide, methotrexate, cyclosporine, mycophenolate mofetil) are still widely used in clinical practice, mostly in association with steroids and/or as steroid-sparing agents, with reported efficacy in 40% to 50% of cases (Table 1).16,24-28 Their long-term detrimental side effects are frequently underestimated, and have never been investigated in prospective randomized-controlled trials.
Patient 3
A 45-year-old woman was diagnosed with wAIHA (Hb, 7.6 g/dL; white blood cells [WBCs], 6.9 × 109/L; normal differential counts and platelets; LDH, 1.9 ULN; reticulocytes, 250 × 109/L; unconjugated bilirubin, 2.8 mg/dL; DAT IgG+) with additional positivity for anti-nuclear antibodies (1/160 titer, homogeneous pattern), anti–β2-glycoprotein 1 IgG, anti-cardiolipin IgG and IgM, and lupus-like anticoagulant (2 positive of 3 tests). Clinical criteria for the diagnosis of a definite systemic autoimmune disease (in particular, systemic lupus erythematosus and antiphospholipid syndrome) were not met. An anti-HBs antibody (Ab) test was positive, with HBsAg, anti-HBc Ab, anti HBe Ab, HBV-DNA, and anti-HCV Ab−. Renal and liver function tests were normal. She was successfully treated with steroids, but relapsed 6 months after discontinuation (Hb, 6.9 g/dL; WBCs, 8.9 × 109/L; lymphocytes, 5.2 × 106/mL; LDH, 2.2 ULN; reticulocytes, 205 × 109/L; unconjugated bilirubin, 2.9 mg/dL; and DAT confirmed positive for IgG alone). Flow cytometry was consistent with the diagnosis of B-cell chronic lymphocytic leukemia (CLL; B-CLL) (stage 0A, negative fluorescence in situ hybridization, unmutated IGHV status). BM evaluation (aspirate and trephine biopsy) showed erythroid hyperplasia, B-CLL infiltration (5% to 10%), and normal cytogenetics. A total-body computed tomography scan was unremarkable. The patient was treated with a standard dose of rituximab; steroid tapering was held at 0.3 mg/kg per day during the 4 weeks of rituximab. A progressive amelioration of Hb and hemolytic markers was observed, with a complete response at month +6 and subsequent progressive steroid withdrawal (month +9). The patient is still in remission after 2 years, and is on “watch-and-wait” follow-up for CLL.
Commentary on patient 3
The first issue arising from this case is whether and when to perform BM evaluation in wAIHA. Although not strictly recommended at diagnosis in primary AIHA, it is advisable at first relapse, in order to identify any underlying conditions, and to confirm a good erythroid reservoir.3,9,13 Moreover, it may be useful in primary forms to examine the prevalent type of lymphocyte infiltrate, to guide immunosuppressive therapy.29 Notably, response to rituximab became evident 1 month from the end of treatment, emphasizing the time needed to achieve a complete response.
It is generally thought that treatment of CLL-associated AIHA should be the same as that for primary disease, provided that CLL requires no treatment.4,9,30 Current and novel specific therapies in the event of AIHA relapse after rituximab are reported in Table 2, including monoclonal antibodies, small molecules, and combination therapies.4,30-47 Splenectomy was excluded given its poor efficacy in CLL-associated forms.9,30 Further concern about this option is its known thrombotic risk (10% to 20% of AIHA cases), which may have been particularly high in patient 3 due to antiphospholipid positivity. Reported thrombotic events in AIHA may be severe, and include pulmonary emboli, splanchnic thromboses, stroke, and cardiac ischemia. In addition to splenectomy, active disease (high LDH and low Hb levels) is associated with an increased risk.48 Prophylactic anticoagulation may be considered, and is highly recommended in the presence of additional risk factors (age, >70 years; reduced mobility; acute infection; previous thrombosis; preexisting thrombophilic condition; recent trauma and/or surgery; heart and/or respiratory failure; active cancer).1,8,9,16
With patient 3, one more reason to avoid splenectomy was positive serology for systemic autoimmune conditions, which may precede overt disease. In fact, splenectomy has poor effectiveness in AIHA secondary to systemic autoimmune disorders and immunodeficiencies.49 Recently, the availability of molecular tools has increased the chances of identifying these forms early: mutations in KDM6A or KMT2D have been identified in Kabuki syndrome, and mutations in genes implicated in primary immunodeficiencies (TNFRSF6, CTLA4, STAT3, PIK3CD, CBL, ADAR1, LRBA, RAG1, and KRAS) have been reported in 40% of Evans syndrome patients, an association of AIHA and ITP.4,50 Whether these findings might influence therapies is an exciting subject for future investigation.
Finally, there is increasing awareness of underlying subclinical or chronic infections that may be reactivated by immunosuppressive therapies. Other than lamivudine for HBV, no precise indications are available for other traditional treatments (steroids), newer therapies (complement, proteasome, and kinases inhibitors), or other infections (tuberculosis, HCV, herpes viruses, and others).1,3,9 Future studies are needed, and a case-by-case discussion with an infectious disease expert is advisable in case of clinical suspicion and/or heavily treated patients.
Are R/R cases of wAIHA always “pure” AIHAs?
The mechanisms involved in the occurrence of autoimmune diseases, and their subsequent evolution into chronic or relapsing disorders, are not completely understood, but involve genetic background, environmental factors, and dysregulation of the complex immunologic pathways and effectors involved in the recognition of self and nonself. Focusing on AIHA, autoimmunity is often triggered by molecular mimicry between self-antigens and foreign antigens for diseases associated with bacterial, mycoplasma, or viral infections.3 Likewise, neoantigens generated by drugs/chemicals may be responsible for triggering drug-induced AIHA.51,52
Finally, it is worth citing the emergence of “forbidden clones” in AIHA secondary to lymphoproliferative disorders, which may be an important factor for relapse and chronicity. Regarding autoimmunity against BM, aplastic anemia is a typical example, being caused by an autoimmune attack against hematopoietic stem cells. Moreover, there is evidence that autoimmune phenomena are present in several conditions other than overt and “classic” autoimmune diseases, such as myelofibrosis and myelodysplasia.53,54 In particular, the presence of anti-erythroblast antibodies, an overinflammatory cytokine milieu, and a proapoptotic pattern have been reported in 50% of patients with low-risk MDS, suggesting a possible pathogenic role in the ineffective erythropoiesis and an underlying unfavorable marrow microenviroment.54
Patient 4
A 69-year-old woman has been treated at our institution since 2009. She was initially diagnosed with wAIHA IgG+ and primary biliary cirrhosis with positive anti-mitochondrial antibodies. She has been treated twice with steroids, and (following a relapse) with low-dose rituximab, first obtaining a complete response, but thereafter only a partial one. In 2010, BM evaluation showed mild dysplasia, markers of hemolysis and DAT became negative, and the patient required occasional transfusion support. Recombinant erythropoietin (EPO; rEPO) was started, with amelioration of anemia until 2011, when worsening pancytopenia developed. A reevaluation of marrow histology proved consistent with aplastic anemia. She was treated, in 2012 and 2013, with 2 cycles of antithymocyte globulin (rabbit and horse, respectively) plus cyclosporine and steroids, obtaining a partial response. Since 2015, the patient has become transfusion dependent, and danazol treatment was unsuccessful. BM reevaluation (2018) showed 20% cellularity with 10% T-lymphocyte infiltration, normal karyotype, and no evidence of overt MDS. She is currently RBC transfusion dependent, and on therapy with eltrombopag (platelets, ∼70 × 109/L) and iron chelation.
Commentary on patient 4
The case presented is paradigmatic of possible evolution from a predominant autoimmunity against the “periphery” (erythrocytes), as observed in AIHA, to a self-attack vs the “center” (BM precursors) typical of aplastic anemia, with features of myelodysplasia in between. Myelodysplasia has multiple declinations and overlaps: low-risk MDS, hypoplastic MDS, idiopathic cytopenia of uncertain significance (ICUS), idiopathic dysplasia of uncertain significance (IDUS), and clonal cytopenias of indeterminate potential.55,56 Some may speculate that a careful revision of BM biopsies and screening for MDS or BMF-associated mutations would probably reallocate several R/R AIHA cases to the ICUS/IDUS category.57,58 In fact, approximately one-third of AIHAs show BM reticulinic fibrosis, which correlates with higher rates of relapse, dyserythropoietic BM features, and increased levels of transforming growth factor β (TGF-β), a well-known inhibitory cytokine.29 Re-evaluation of R/R AIHA cases is recommended, as they may display clinical and immunological features of ICUS/IDUS; in particular, it may be necessary to retailor therapies, avoiding excess immunosuppression or shifting to new immune-modulating agents.
Indeed, rEPO administration, which is known to be effective in treating MDS, has been useful for some time in patient 4’s treatment. More generally, rEPO has recently been reported as effective in 70% of both primary and secondary AIHA patients, with a median Hb increase of 2.4 mg/dL (range, 2-83 mg/dL), independent of AIHA type and number of previous therapy lines.59 The latter finding suggests no dysplasia-inducing effect from multitreatment, although this should be addressed in larger studies. Moreover, endogenous EPO levels were reduced in AIHA, and correlated with ineffective erythropoiesis and inflammatory cytokine tumor necrosis factor α levels. The patient was also treated with danazol, a drug which has received recent attention for its use in BMF treatment, and telomeropathies.60-62 Finally, whether the presence of BMF and MDS-associated mutations (SF3B1, SRSF2, TET2, DNMT3A, ASXL1, BCOR/BCORL1, TP53, RUNX1, PIGA) plays a role in the described evolution will be a fascinating area to explore in the future.13
What to do in acute and severe cases?
Acute and severe cases represent a challenge for clinicians, even those familiar with AIHA. Usually there is no time to test the efficacy of different treatments, and rapidly worsening organ failures lead to a burden of overlapping therapies. In such cases, mortality rates of up to 57% have been reported, despite intensive treatment, including transfusions, steroid boluses, IV immunoglobulins (IVIgs), rituximab, rEPO, and plasma exchange (PEX).16,28,63,64 Likewise, a recent study of 44 AIHA cases admitted to the intensive care unit for severe anemia reported 30% mortality.65 Associated conditions may also impact on disease severity and prognosis, with higher mortality reported in AIHA secondary to systemic lupus erythematosus, lymphoma, and cancer.3,9 Transfusion issues may also create challenges in managing AIHAs in an acute setting, as the principle “never deny but avoid if unnecessary” is not always easily applicable to transfusion decisions. Moreover, the presence of alloantibodies (reported in 15% to 40% of AIHA cases) may complicate and delay the selection of compatible units, as well as fuel acute hemolysis and/or increase delayed posttransfusion reactions.3,9,14
Patient 5
A 31-year-old man presented at the emergency department in acute and severe wAIHA crisis (Hb, 2.8 g/dL; reticulocytes, 57 × 109/L; unconjugated bilirubin, 4.9 mg/dL; LDH, 3.5 ULN; DAT+, IgG plus C), along with fever (102°F), tachycardia (105 bpm), tachypnea (24 bpm), leukocytosis (16.3 × 109/L), and hemoglobinuria. His past history was unremarkable except for an episode of ITP 5 years before admission, successfully treated with steroids. Platelet counts, and liver and kidney function tests, were within the normal range. A whole-body computed tomograpy scan was negative for splenomegaly or lymph node enlargement, thrombosis, and infectious lesions. Normal coagulation parameters and absence of schistocytes excluded disseminated intravascular coagulation and other microangiopathies. The patient was transfused and treated with steroid boluses (methylprednisolone 250 mg IV daily for 3 days), IV antibiotics, and prophylactic heparin. Due to his septic state, IVIgs were started on day +3. Hb levels stabilized around 4 g/dL with a further 3 blood units, but reticulocytopenia and elevated LDH persisted. On day +5 the patient developed acute renal failure (creatinine, 4 mg/dL), and was transferred to the intensive care unit. The patient received fluids and vasopressors with amelioration of renal function, and underwent 3 daily plasma exchanges. Despite steroid treatment, anemia and reticulocytopenia did not significantly improve (day +10). After exclusion of underlying conditions at BM evaluation, rituximab standard dose was started. On day +17, rEPO (epoietin α, 40 000 IU per week) was added, with progressive amelioration of Hb values (8-10 g/dL) and the appearance of the reticulocyte crisis. After discharge, the patient continued steroid therapy with tapering and was able to discontinue rEPO after 6 weeks.
Commentary on patient 5
Patient 5 had been diagnosed with Evans syndrome, with very severe/acute AIHA, and previous ITP (known to occur together or separately). Profound autoimmunity was present, sustained by autoantibodies that activate complement, causing intravascular hemolysis, and associated with reticulocytopenia, possibly due to antibodies against erythroblasts that hamper BM compensation.3,16,28,66 The various therapies administered were directed at various pathogenetic mechanisms: steroids and rituximab to decrease inflammation and antibody-producing B lymphocytes, IVIg to mask ADCC and help against concomitant infection, PEX to remove excess immune mediators, and rEPO to overcome a “shocked” BM.59
Rituximab is usually recommended as a second-line therapy; however, in urgent situations, the exact time of steroid failure is not easy to define. In AIHAs following hematopoietic stem cell transplantation, which are generally severe, refractory to steroids, and fatal, rituximab is recommended either as a frontline or early second-line therapy.67-70 This has led to improved outcomes, suggesting that this approach might work in all acute settings. PEX is clearly a temporizing measure until specific treatments become effective. Although evidence for its efficacy is limited and controversial, the removal of pathogenic immune complexes, circulating autoantibodies, and activated complement may be useful in acute and rapidly deteriorating cases.
The exact mechanism by which IVIg acts is not fully understood.7,9,71 Recently, it has been suggested that IVIg administration can determine saturation of the neonatal Fc receptor (FcRn), which is responsible for the salvage of IgG from catabolism, thus resulting in accelerated clearance of endogenous IgG (including pathogenic autoantibodies). New drugs that block FcRn are under development, hopefully providing a PEX-like strategy for the acute setting in the future. Likewise, complement inhibitors that are close to being approved for cold agglutinin disease (CAD; where complement activation is the main pathogenic mechanism) are under development in wAIHA and are likely to be useful in IgG-plus-C forms.7
Figure 2 summarizes the different approaches in acute and very severe cases, compared with nonsevere ones, including options beyond second-line therapy. In fact, patient 5 is at risk of further relapse, given the AIHA type and severity, and the diagnosis of Evans syndrome. Hazard ratios for relapse increased from 1.61 to 1.74 and 1.98 for Hb values 8.1 to 10 g/dL, 6.1 to 8 g/dL, and <6 g/dL, respectively; also, taking into account Hb as a continuous variable, each gram of reduction resulted in 7% higher relapse risk.28 Moreover, Evans syndrome, acute renal failure, and infection (all present in our patient) have been associated with increased mortality (hazard ratios of 8, 6.3, and 4, respectively).
Therefore, it is essential to plan a future therapeutic strategy. Splenectomy, provided patient consent, is a valid option, but it is prudent to wait ∼1 year from diagnosis before adopting this irreversible and not definitively curative option. Rituximab has already been used, and retreatment, although feasible, would probably be ineffective in the event of an early relapse (within 8-12 months of diagnosis). Meanwhile, the patient might be managed with steroids and classic immunosuppressants as steroid-sparing agents (Table 1). Among future therapeutic options, enrollment in a clinical trial is highly advisable. Table 3 shows new treatments and ongoing studies with tyrosine kinases, complement, proteasome, FcRn, and ADCC inhibitors, as well as case reports describing efficacy of daratumumab, sirolimus, and abatacept.13,72-86
Conclusion
wAIHA is a greatly heterogeneous disease with an unpredictable clinical course, due to its multiple immunologic mechanisms, and their variable role over time. This unpredictability should always be considered, and re-evaluation of DAT, hemolytic parameters, and BM characteristics is recommended, particularly in R/R cases. Complete BM evaluation is recommended for patients with suspected underlying disease or those who have relapsed after first-line steroid therapy. Consequently, therapies should ideally be used to target the prevailing immunopathogenic mechanism (Figure 3). Our main clinical recommendations include proper use of steroids (not for <6 months, including tapering; never for >1 year), early use of rituximab or even retreatment, careful consideration of splenectomy at an appropriate time, and accurate detection of a possible evolution to ICUS/IDUS in R/R patients. Likewise, careful investigation for secondary forms is highly advisable, as this may change the therapeutic strategy quite considerably. Given the lack of evidence beyond the second line, and the decline of splenectomy, enrollment in clinical trials should be pursued strongly in R/R wAIHA cases. As new target therapies become available, identifying the optimal choice, sequence, and combination of drugs, with the aim of “curing” wAIHA, will be an increasingly complex but fascinating challenge.
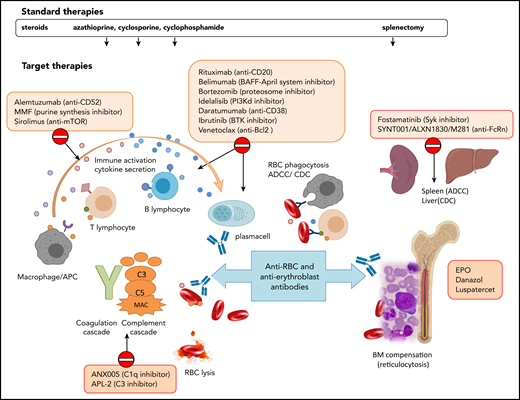
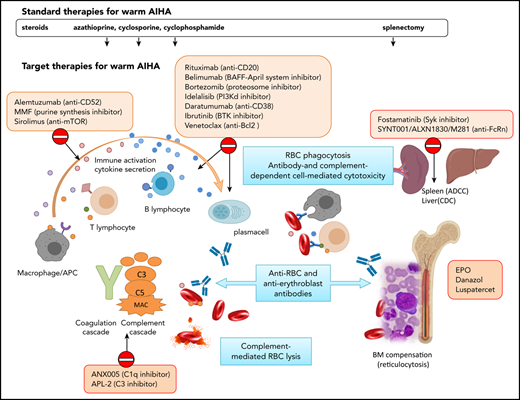
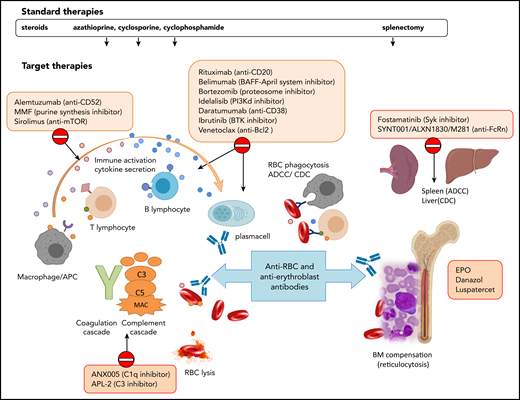
Standard and target therapies for wAIHA. This figure represents the various immunologic mechanisms involved in AIHA pathogenesis, including macrophages, T and B lymphocytes, cytokines, activation of the complement cascade, ADCC in the spleen, and/or complement-dependent cytotoxicity (CDC) in the liver, and lack of BM compensation. Standard therapies include steroids and immunosuppressors that do not act specifically on the various mechanisms and splenectomy. Target therapies are directed against specific immunological mechanisms. APC, antigen-presenting cell; BAFF, B-cell–activating factor; BTK, Bruton tyrosine kinase; CAD, cold agglutinin disease; MAC, membrane attack complex; MMF, mycophenolate mofetil; mTOR, mammalian target of rapamycin; PI3K, phosphoinositide 3-kinase; Syk, spleen tyrosine kinase.
Standard and target therapies for wAIHA. This figure represents the various immunologic mechanisms involved in AIHA pathogenesis, including macrophages, T and B lymphocytes, cytokines, activation of the complement cascade, ADCC in the spleen, and/or complement-dependent cytotoxicity (CDC) in the liver, and lack of BM compensation. Standard therapies include steroids and immunosuppressors that do not act specifically on the various mechanisms and splenectomy. Target therapies are directed against specific immunological mechanisms. APC, antigen-presenting cell; BAFF, B-cell–activating factor; BTK, Bruton tyrosine kinase; CAD, cold agglutinin disease; MAC, membrane attack complex; MMF, mycophenolate mofetil; mTOR, mammalian target of rapamycin; PI3K, phosphoinositide 3-kinase; Syk, spleen tyrosine kinase.
Acknowledgment
This work was supported by research funding from Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico (RC 2018).
Authorship
Contribution: W.B. and B.F. conceived and wrote the manuscript.
Conflict-of-interest disclosure: W.B. provided consultancy services to Agios, Alexion, Apellis, Biocryst, Bioverativ, Incyte, Momenta, and Novartis, and received lecture fees/congress support from Alexion, Incyte, Novartis, and Sanofi. B.F. provided consultancy services to Apellis, Momenta, and Novartis, and received lecture fees/congress support from Alexion and Apellis.
Correspondence: Wilma Barcellini, Hematology, Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico, Via F. Sforza 35, 20122 Milan, Italy; e-mail: wilma.barcellini@policlinico.mi.it.