

TO THE EDITOR:
T-cell acute lymphoblastic leukemia (T-ALL) and T-cell lymphoblastic lymphoma (T-LBL) are aggressive neoplasms that result from the proliferation of T-lymphoid progenitors blocked at thymic stages of differentiation. They account for 15% and 25% of pediatric and adult ALLs, respectively. T-ALL/T-LBL are associated with a wide range of acquired genetic abnormalities that contribute to developmental arrest and abnormal proliferation.1,2 Although intensive treatment protocols have markedly improved the outcomes of children with T-ALL, cure rates remain below 60% for adults and 85% for children.3-5 The prognosis is particularly poor in relapsing patients, highlighting an urgent need for risk stratification factors at diagnosis.6,7
The IKAROS transcription factor (encoded by the IKZF1 gene on chromosome 7p12.2) is a member of the zinc finger family of DNA-binding proteins that acts as a critical regulator of hematopoiesis and lymphoid differentiation.8 IKZF1 is recurrently affected by various genetic alterations in B-cell acute lymphoblastic leukemia (B-ALL). Genomic alterations in IKZF1 are found in ∼15% of childhood B-ALL cases and in 40% of adult B-ALL cases, with a higher incidence in poor prognosis cases, including BCR-ABL1 (70%) or BCR-ABL1–like (40%) B-ALL.9,10 Of note, the IKZF1 alteration consistently exhibited its poor prognostic impact in B-ALL, and clinical trials increasingly integrate IKZF1 gene status in risk stratification algorithms.11,12
In contrast, both the incidence and prognostic influence of IKZF1 alterations in T-ALL/T-LBL are poorly characterized.13 To specify the role of IKZF1 alterations in T-ALL/T-LBL, we conducted a comprehensive analysis using pan-exon deep sequencing of 1260 adult and pediatric T-ALL/T-LBL patients (supplemental Figure 2, available on the Blood Web site), including 980 T-ALL cases and 280 T-LBL cases. Diagnostic DNA samples were analyzed by using an 80-gene pan-exon capture-panel (details included in the supplemental Methods). IKZF1Alt screening was performed by computational approaches previously described for the detection of copy number variants from next-generation sequencing data.14 IKZF1 deletions were confirmed with multiplex ligation-dependent probe amplification (MLPA) analysis and/or microarray-based comparative genomic hybridization (array CGH). Patient protocols and clinical trials,3,15,16 immunophenotypic and molecular characterization of T-ALL and T-LBL samples, minimal residual disease (MRD) assessment, gene mutation screening, array CGH, MLPA, statistical analysis, and additional details are included in the supplemental Methods.
IKZF1 mutations were identified in 42 cases, including 33 (3.4%) of 980 T-ALL cases and 9 (3.2%) of 280 T-LBL cases (Figure 1A). The majority of mutations were missense (24 of 42 [60%]) within a mutational hotspot in exon 5 affecting amino acid p.N159 (N159S/T) located in the DNA-binding domain. Interestingly, this mutation was recently described in a new combined immunodeficiency syndrome with potential risk of T-ALL predisposition.17,18 We also detected frameshift or nonsense mutations (17 of 42 [40%]) affecting exons 3-8 and predicted to truncate the protein before the C-terminal dimerization domain, resulting in haploinsufficiency. IKZF1 deletions were detected in 40 cases (3%) including 37 (3.8%) of 980 and 3 (1.5%) of 280 in T-ALL and T-LBL, respectively (Figure 1B). All were confirmed by MLPA and/or array CGH (supplemental Figure 2; supplemental Figure 3A-B). Of note, most cases (30 of 40 [75%]) harbored pan-genic deletions (exons 1-8), leading to haploinsufficiency; only 10% (4 of 40) intragenic deletions (exons 4-7), predicted to induce a dominant-negative effect, were observed. This suggests that, in contrast to B-cell precursor-ALL (BCP-ALL),12 the main consequences of IKZF1 deletions in T-ALL would be haploinsufficiency rather than a dominant-negative effect. IKZF1 deletions and mutations were mutually exclusive, and no biallelic inactivation of IKZF1 was observed, suggesting that residual IKZF1 activity may be required for T-lineage leukemogenesis. Overall, IKZF1Alt was identified in 82 (6.5%) of 1260 T-ALL/T-LBL cases (7.1% T-ALL and 4.3% T-LBL) and were more frequent within adult T-ALL/T-LBL cases compared with pediatric cases (54 of 699 adult T-ALL/T-LBL compared with 28 of 561 pediatric cases; P = .05) (supplemental Figure 2). The oncoplot highlighting the main comutations observed and mutations within individual IKZF1Alt cases are reported in supplemental Table 3 and supplemental Figure 4.
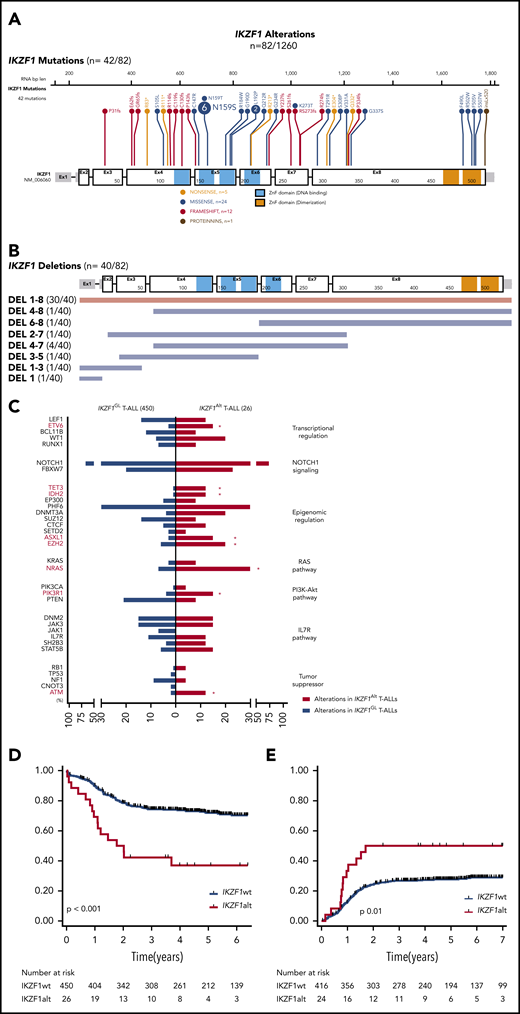
IKZF1 mutational and deletion patterns according to patient occurrence. (A) Gene map describing IKZF1 intragenic mutational patterns according to patient occurrence. (B) Gene map describing IKZF1 deletion patterns according to patient occurrence. (C) Genetic profiles of IKZF1Alt T-ALL in the FRALLE and GRAALL 03-05 protocols. Comparison of mutational profiles according to pathways between IKZF1Alt T-ALLs (n = 26) and IKZF1GL T-ALLs (n = 450), with a focus on alterations found in at least 5% of the whole cohort. Percent frequencies in each group are indicated. Genes are grouped according to functional categories. OS (D) and cumulative incidence of relapse (E) in FRALLE and GRAALL treated patients. *P < .05. IL7R, interleukin-7 receptor; PI3K-Akt, phosphatidylinositol 3-kinase/protein kinase B.
IKZF1 mutational and deletion patterns according to patient occurrence. (A) Gene map describing IKZF1 intragenic mutational patterns according to patient occurrence. (B) Gene map describing IKZF1 deletion patterns according to patient occurrence. (C) Genetic profiles of IKZF1Alt T-ALL in the FRALLE and GRAALL 03-05 protocols. Comparison of mutational profiles according to pathways between IKZF1Alt T-ALLs (n = 26) and IKZF1GL T-ALLs (n = 450), with a focus on alterations found in at least 5% of the whole cohort. Percent frequencies in each group are indicated. Genes are grouped according to functional categories. OS (D) and cumulative incidence of relapse (E) in FRALLE and GRAALL treated patients. *P < .05. IL7R, interleukin-7 receptor; PI3K-Akt, phosphatidylinositol 3-kinase/protein kinase B.
We then investigated the clinical characteristics linked to IKZF1Alt in a subset of 476 patients, including 215 adults enrolled in the GRAALL-2003/05 trials and 261 children enrolled in the FRALLE-2000 trial (GRAALL-2003, #NCT00222027; GRAALL-2005, #NCT00327678) (Table 1; supplemental Methods). Diagnostic peripheral blood or bone marrow samples from 1258 adults and children with T-ALL or T-LBL were analyzed after informed consent was obtained at diagnosis according to the Declaration of Helsinki. The incidence of IKZF1Alt in this cohort was 5.5% (26 of 476), including 16 deletions and 10 mutations. IKZF1Alt were observed in 7% of adults and 4.2% of children (P = .2), with a median age slightly higher in IKZF1Alt cases (23.5 years vs 15.2 years; P = .1). IKZF1Alt was associated with an immature immunophenotype (47% vs 20%; P = .02). In line with this finding, IKZF1Alt correlated positively with abnormalities known to be associated with an immature phenotype, including K/N-RAS mutations (31% vs 9%; P = .003), EZH2 (5 of 26 [20%] vs 27 of 450 [6%]; P = .02), ASXL1 (4 of 26 [15%] vs 15 of 450 [3%]; P = .02), ETV6 (4 of 26 [15%] vs 13 of 450 [3%]; P = .01), and DNMT3A (5 of 26 [20%] vs 17 of 450 [4%]; P = .045) mutations (Figure 1C). Conversely, IKZF1Alt were virtually exclusive, with SIL-TAL1+ and PTEN altered cases known to be associated with a mature T-cell receptor-αβ lineage.
In BCP-ALL, IKZF1Alt are enriched in the high-risk subgroup of Ph+ BCP-ALL and recently characterized by the presence of a gene expression profile similar to Ph+ ALL but lacking the canonical BCR-ABL1 fusion, therefore named BCR-ABL1-like or Ph+-like ALL.9,19 Importantly, Ph+-like signature is virtually absent in T-ALL.20 In contrast, here we observed that IKZF1Alt are associated with epigenetic mutations/deletions.
Interestingly, IKZF1-deficient mice develop T-cell malignancy with high penetrance, highlighting the suppressor function for IKAROS in T-cell lineage.21 Furthermore, in murine T-cell leukemogenesis, IKAROS directly cooperates with NOTCH1 activation to promote leukemia.22,23 This crosstalk between NOTCH1 and IKAROS could explain the discrepancy observed concerning the pattern of IKZF1Alt in T-ALL vs Ph+-like BCP-ALL, and it led us to suspect a specific oncogenic mechanism associated with IKZF1Alt in T-ALL requiring further investigations.
IKZF1Alt cases did not differ significantly with regard to sex, white blood cell count, central nervous system involvement, and prednisone response (Table 1). Although IKZF1Alt did not affect the complete remission rate, patients with IKZF1Alt were more likely to have a positive postinduction MRD (10−4 threshold, 67% vs 34%; P = .01). Patients with IKZF1Alt had an inferior outcome compared vs those with IKZF1GL, with a higher cumulative incidence of relapse (5-year cumulative incidence of relapse, 50% vs 28%; specific hazard ratio, 2.12; 95% confidence interval [CI], 1.17-3.86) and a shorter overall survival (OS) (5-year OS, 37% vs 73%; hazard ratio, 2.94; 95% CI, 1.74-4.96) (Figure 1D-E). This prognostic impact was observed in both pediatric and adult cohorts (supplemental Figure 5A-D). Of note, the 10 IKZF1-mutated and 16 IKZF1-deleted cases exhibited comparable clinico-biological features and were associated with worse prognosis compared with IKZF1GL cases (supplemental Table 4; supplemental Figure 6). In multivariate analysis, considering variables associated with OS in univariate analyses as covariates, IKZF1Alt remained significantly associated with a shorter OS, even after inclusion of postinduction MRD in the model (supplemental Table 5). It is noteworthy that the prognostic impact of IKZF1Alt status was also observed after adjustment on the 4-gene NOTCH1/FBXW7/RAS/PTEN classifier and postinduction MRD, which identified poor prognosis patients in both GRAALL and FRALLE trials.3,4
In conclusion, we describe IKZF1Alt among 1260 children and adults with immature T-ALL/T-LBL and define for the first time its frequency and, importantly, its poor outcome in T-ALL in multivariate models. IKZF1Alt should be considered as a significant prognosis marker in addition to MRD and the 4-gene oncogenetic classifier to predict poor outcomes in T-ALL.
E-mail the corresponding author for original data.
The online version of this article contains a data supplement.
Acknowledgments
The authors thank all participants in the GRAALL-2003 and GRAALL-2005 study groups, the Société Française de lutte contre les Cancers et les leucémies de l'Enfant et de l'Adolescent (SFCE), and the investigators of the 16 SFCE centers involved in collection and provision of data and patient samples. They also thank V. Lheritier for collection of clinical data.
The GRAALL was supported by grants P0200701 and P030425/AOM03081 from the Programme Hospitalier de Recherche Clinique, Ministère de l’Emploi et de la Solidarité in France, and the Swiss Federal Government in Switzerland. Samples were collected and processed by the AP-HP “Direction de Recherche Clinique” Tumor Bank at Necker-Enfants Malades. M.S. was supported by “Soutien pour la formation à la recherche translationnelle en cancérologie dans le cadre du Plan cancer 2009-2013.” This work was supported by grants to Necker laboratory from the “Association Laurette Fugain,” Association pour la Recherche contre le Cancer (Equipe Labellisée), Institut National du Cancer PRT-K 18-071, and the Fédération Leucémie espoir and Horizon Hemato.
Authorship
Contribution: N.B., V.A., and M.S. conceived the study and oversaw the project; M.S., M.-E.D., L.L., E.L., C.G., N.G., J.-M.C., I.A., V.G., N.I., H.D., A.B., A.P., and N.B. provided study materials or patients; M.S., L.L., E.M., and V.A. performed molecular analyses; M.S., L.L., and V.A. collected and assembled data; N.B. and M.S. performed statistical analysis; M.S., L.L., V.A., and N.B. analyzed and interpreted data; M.S., N.B., E.M., and V.A. wrote the manuscript; and all authors approved the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Vahid Asnafi, Laboratory of Onco-Hematology, Tour Pasteur 2eme etage, Necker Enfants Malades Hospital, 149 Rue de Sevres, 75015 Paris, France; e-mail: vahid.asnafi@aphp.fr; or Nicolas Boissel, Hôpital Saint-Louis, Service d’Hématologie Adulte, 1 Avenue Claude Vellefaux 75010 Paris, France; e-mail: nicolas.boissel@aphp.fr.

