Abstract
Stimulator of interferon genes (STING) is an innate immune sensor of cytoplasmic dsDNA originating from microorganisms and host cells. STING plays an important role in the regulation of murine graft-versus-host disease (GVHD) after allogeneic hematopoietic stem cell transplantation (allo-HSCT) and may be similarly activated during other transplantation modalities. In this review, we discuss STING in allo-HSCT and its prospective involvement in autologous HSCT (auto-HSCT) and solid organ transplantation (SOT), highlighting its unique role in nonhematopoietic, hematopoietic, and malignant cell types.
Introduction
Stimulator of interferon genes (STING) is an endoplasmic reticulum protein that acts as an indirect sensor of cytoplasmic double-stranded DNA (dsDNA).1 The sources of DNA that induce cyclic dinucleotides (CDNs) include the genomes of invading pathogens, including herpes simplex virus 1 (HSV-1) and cytomegalovirus, whereas certain bacteria can secrete CDNs (eg, Listeria monocytogenes) after infection of the host.2-4 STING signaling has now been shown to be essential for protecting the cell against a variety of pathogens and even against the development of cancer by promoting antitumor immune responses.5,6 The canonical ligand for STING is the CDN 2′,3′-cGAMP which is produced as a result of DNA sensing by the protein cyclic GMP-AMP synthase (cGAS). STING can also be activated by other CDNs, including cyclic di-AMP (c-di-AMP) and cyclic di-GMP (c-di-GMP).7,8 After binding to CDNs, STING recruits TBK1 and traffics to perinuclear regions.9 Downstream STING signaling results in the activation of the transcription factors interferon regulatory factor 3 (IRF3) and NF-κB, which induce type I interferons (T1IFNs) and additional inflammatory molecules (Figure 1A).10 Notably, STING alleles in the human population have been identified that encode proteins that are largely unresponsive to CDNs, raising the notion that there may be clinical implications for individuals with these genotypes.11 Patients are administered liquid (bone marrow transplants) and solid organ transplants (SOTs) to replace damaged tissue compartments. Continuing efforts to develop clinically useful STING agonists and antagonists may prove effective as therapies to reduce or prevent allograft rejection (host-versus-graft) in SOT and graft-versus-host disease (GVHD) after allogeneic hematopoietic stem cell transplantation (allo-HSCT). Notably, complications in transplant recipients include an array of infections and hematopoietic malignancies, and therefore regulating STING may affect immune defense against microbial pathogens and cancer in such patients. In this review, we focus on current literature regarding STING and its role in allo-HSCT, which is currently the predominant transplantation arena where it has begun to be studied. We also speculate on the involvement of STING in autologous HSCT (auto-HSCT) and SOT and highlight recent work using new STING compounds, which are bringing STING-targeted agents closer to implementation in the clinic.
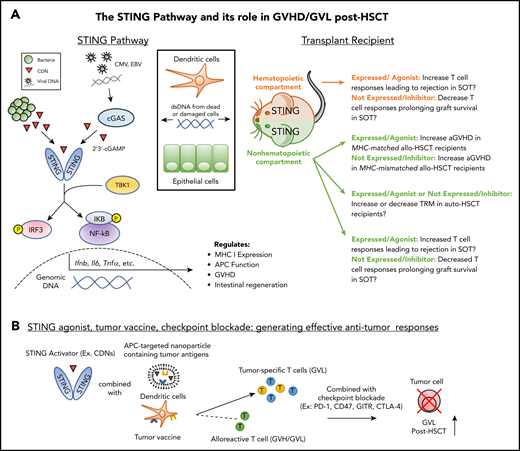
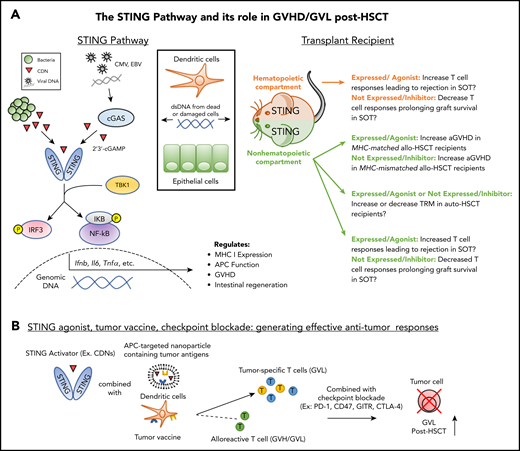
The STING pathway in NH and HCs: prospective contribution to transplant outcome. (A) Left: As a result of transplantation-related damage, dsDNA from host cells (eg, dendritic cells, epithelial cells) or viruses induce cyclic dinucleotide (CDN) production by cGAS which bind to and activate STING. Bacterial CDNs can also activate STING directly. Downstream of STING signaling, activation of IRF3 and NF-κB induce the production of cytokines, which regulate immune activation and reparative processes such as epithelial regeneration. Right: STING activation in the NH or hematopoietic compartments during SOT could increase T-cell responses against donor grafts. STING activation in the NH compartment during allo-HSCT increases aGVHD in MHC-matched transplant recipients; however, expression of STING in this compartment decreases aGVHD in MHC-mismatched transplant recipients. The potential role of STING in auto-HSCT is unclear. Promotion of epithelial repair in the GI tract and/or increased anti-pathogen T-cell responses could diminish recipient transplant-related mortality (TRM). Alternatively, increased cytokine production as a result of conditioning could elevate recipient TRM. (B) STING-targeted agents (see Table 1) can be combined with tumor vaccination and checkpoint blockade to promote tumor-specific responses without exacerbating GVHD. Application of this strategy could be combined with prophylactic GVHD regimens involving posttransplant cyclophosphamide to reduce potential exacerbation of donor antihost alloreactivity in addition to direct cytotoxic effects on the tumor. EBV, Epstein-Barr virus.
The STING pathway in NH and HCs: prospective contribution to transplant outcome. (A) Left: As a result of transplantation-related damage, dsDNA from host cells (eg, dendritic cells, epithelial cells) or viruses induce cyclic dinucleotide (CDN) production by cGAS which bind to and activate STING. Bacterial CDNs can also activate STING directly. Downstream of STING signaling, activation of IRF3 and NF-κB induce the production of cytokines, which regulate immune activation and reparative processes such as epithelial regeneration. Right: STING activation in the NH or hematopoietic compartments during SOT could increase T-cell responses against donor grafts. STING activation in the NH compartment during allo-HSCT increases aGVHD in MHC-matched transplant recipients; however, expression of STING in this compartment decreases aGVHD in MHC-mismatched transplant recipients. The potential role of STING in auto-HSCT is unclear. Promotion of epithelial repair in the GI tract and/or increased anti-pathogen T-cell responses could diminish recipient transplant-related mortality (TRM). Alternatively, increased cytokine production as a result of conditioning could elevate recipient TRM. (B) STING-targeted agents (see Table 1) can be combined with tumor vaccination and checkpoint blockade to promote tumor-specific responses without exacerbating GVHD. Application of this strategy could be combined with prophylactic GVHD regimens involving posttransplant cyclophosphamide to reduce potential exacerbation of donor antihost alloreactivity in addition to direct cytotoxic effects on the tumor. EBV, Epstein-Barr virus.
STING signaling in recipient cells affects allo-HSCT transplant outcome
Despite the widespread use of allo-HSCT for more than 5 decades, GVHD that is driven by major and minor histocompatibility antigen disparities between donor and recipient remains a major cause of nonrelapse morbidity and mortality.12-14 GVHD is potentiated when innate immune sensors, mainly in hematopoietic and nonhematopoietic antigen-presenting cells (APCs), are activated in response to pathogen-associated molecular patterns and damage-associated molecular patterns released by pretransplant chemotherapy or irradiation that result in upregulating certain cytokines.15 Recent work by our laboratory and others has established that 1 such innate sensor, STING, is a potent regulator of GVHD.16,17 The initial studies that demonstrated the ability of STING to regulate GVHD used murine models of major histocompatibility complex (MHC)–mismatched HSCT and recipients (B6-Goldenticket, B6-STINGgt/gt) expressing a missense mutation in the STING gene.18 Transplants into B6-STINGgt/gt recipients resulted in higher lethality compared with B6 wild-type recipients, which has been attributed to the reduced ability of STING-deficient recipients to regenerate their intestinal epithelium after pretransplant conditioning.16
Recently, by using an independently derived STING-deficient mouse (B6-STING−/−), the initial report that murine STING deficiency results in worse GVHD after MHC-mismatched allo-HSCT was independently corroborated.17 These studies also examined MHC-matched HSCT and discovered that STING deficiency ameliorated GVHD in these matched unrelated donor models.17 Donor CD4+ or CD8+ T cells often predominate in preclinical graft-versus-host responses (ie, CD4+ in MHC-mismatched vs CD8+ in MHC-matched).19 Subset studies revealed that STING deficiency protected against GVHD when only donor CD8+ T cells were transplanted into either MHC-mismatched or -matched recipients, thereby reconciling the seemingly disparate results in these models. Interestingly, T1IFN was also shown to differentially regulate CD4+ vs CD8+ T-cell–mediated GVHD.20 Moreover, T1IFN enhanced MHC class I expression on recipient hematopoietic APCs, which are critical for the induction of GVHD.17 In contrast, hematopoietic APCs responding to T1IFNs have diminished allogenicity, suggesting a protective role for STING in MHC-mismatched GVHD.21
Nonhematopoietic (NH) cells are important for the initiation of gut GVHD.22 Hematopoietic APCs seem to play a role after STING signaling following allo-HSCT, and STING expression in NH cells protected against CD4+ T-cell–mediated GVHD but enhanced CD8+ T-cell–mediated GVHD regardless of the allo-HSCT model.17 The importance of the NH compartment was also demonstrated in knockout mice that lacked mitochondrial antiviral-signaling protein, a critical molecule involved in cytoplasmic RNA sensing.16 The specific NH cell types in which STING expression is important for the regulation of GVHD remains unknown, but STING activation in NH cells has been shown to be responsible for the initiation of autoimmune disease involving activated T cells.23 We speculate that in non-hematolymphoid tissue compartments, STING activation within NH-APCs leading to production of T1IFNs and inflammatory cytokines could target parenchymal populations that are distinct from those in the hematopoietic tissues. For example, IFNs could upregulate MHC class I on tissue-resident cells which would alter their susceptibility as target populations. NH-APC cytokine/chemokine (eg, MCP-1/CCL2) production downstream of T1IFNs could promote the trafficking of hematopoietic cell (HC)-APCs into lymphoid tissues thus augmenting alloreactive donor T-cell responses. NH-APCs after HSCT could engage CD8+ effector cells and activate resident memory populations. Activation of the STING pathway in any recipient cell could occur via several routes, depending on the host compartment. After conditioning involving chemotherapy and irradiation, virtually all cells could activate STING in response to dying cells via uptake of dsDNA.24,25
Intestinal microbiota may also play a role in activating STING in recipient cells after HSCT, because pretransplant conditioning directly induces intestinal permeability, which provides exposure of bacterial dsDNA to numerous cell populations.26,27 Consistent with such a pathway, gut microbiota are capable of initiating lethal GVHD through upregulation of MHC class II on NH intestinal epithelial cells.22 Therefore, the extent to which conditioning regimens damage the gastrointestinal (GI) tract may correlate with the level of STING activation in this compartment. In addition, recent studies have reported emerging pathways and new mechanisms leading to STING activation involving noncanonical signaling complexes and micronuclei in damaged cells.28-30 Further studies are required to identify the precise mechanisms by which recipient STING is activated before and during HSCT to better understand how STING signaling differentially regulates GVHD. It should be noted that preclinical mouse studies are typically performed under specific pathogen-free conditions and thus do not mimic the opportunistic viral (eg, reactivation of cytomegalovirus), bacterial, and fungal infections that frequently occur in allo-HSCT patients and can cause mortality (Figure 1A).31-33 Diminishing or augmenting STING signaling could alter the course of such infections, which can influence GVHD. For example, decreasing STING signaling could diminish responses against such microbial pathogens. Alternatively, augmenting STING signaling could enhance anti-pathogen immunity thereby preventing pathogen promotion of GVHD.34,35 Overall, both the timing of STING activation and the transplant parameters will have an impact on how this pathway regulates GVHD to ultimately enable STING agonists and/or antagonists to benefit patients receiving allo-HSCTs.
STING in tumor cells: friend or foe in transplant recipients?
Allogeneic HSCT is most often used to treat hematologic malignancies, primarily leukemias, to exploit the accompanying antitumor (graft-versus-leukemia [GVL]) response. Patients who develop acute GVHD (aGVHD) or chronic GVHD (cGVHD) have a lower frequency of leukemia relapse because of immune activation against alloantigens expressed on both normal and malignant cells.36 STING agonists can promote GVHD,17 and therefore this pathway could augment beneficial GVL responses by facilitating the activation of donor T cells that target alloantigens and tumor antigens (Figure 1B). The outcome of STING activation can reportedly be either immunogenic or tolerogenic. Thus, it is possible that the former would result in increased graft-versus-host and GVL responses, whereas the latter may diminish these responses.37 Alternatively, STING antagonists could diminish GVL by decreasing donor alloantigen-specific or tumor-specific T cells resulting in increased risk of relapse. Several studies have demonstrated that STING-activating agents can improve antitumor T-cell responses in both solid and liquid tumor models.38-40 In solid tumors, in which the majority of STING-related cancer research has been performed, defective STING signaling in tumors has been shown to correlate with increased tumorigenesis in colorectal carcinoma and ovarian cancer.41,42 Interestingly, tumor-derived 2′,3′-cGAMP can trigger STING-mediated T1IFN in neighboring cells as long as cGAS is functional through mechanisms involving the transfer of CDNs from one cell to another.43
STING signaling in tumor cells also correlates with their antigenicity and immunogenicity.44,45 Increased tumor cell immunogenicity can result from tumor-derived DNA, which activates STING signaling in both tumor and dendritic cells (DCs) and leads to the production of T1IFN and increased antitumor immunity.46 STING signaling in solid tumors can also be initiated by irradiation-induced DNA damage, and liquid tumors may be similarly activated in response to pre-HSCT chemotherapy.47 Notably, the dose of irradiation can regulate this response, because higher doses attenuate STING activation as a result of the upregulation of 3′ repair exonuclease 1 (TREX1), which directly competes with cGAS by degrading cytoplasmic dsDNA.48 It is therefore likely that the precise conditioning regimen used for HSCT is in part responsible for the level of STING signaling in response to chemotherapy and irradiation. STING signaling reportedly slows tumor proliferation via control of NF-κB and p53-driven activation of p21.49 In contrast, STING activation has also been shown to be immunosuppressive via the recruitment of myeloid-derived suppressor cells to the tumor site via CCR2.50 Thus, STING signaling in tumors is complex, and in transplant recipients, downstream effects of STING activation are likely to be highly dependent upon both the type of tumor and the conditioning regimen.
STING in donor cells: GVHD and cellular therapy in allo-HSCT recipients
Unfortunately, primary disease is still the most common cause of mortality after allo-HSCT.51 Activation of STING in donor T cells can induce strong antitumor responses despite the antiproliferative effects of T1IFN.52,53 Moreover, STING-induced T1IFN is effective at promoting natural killer cell antitumor activity.54 Strategies to combine HSCT with posttransplant cellular therapy are being developed that may improve transplant outcomes.55 Notably, STING in DCs is reportedly activated by tumor mitochondrial DNA and can be enhanced by tumor CD47 blockade.56 On the basis of such types of findings, it may be noteworthy that pulsed DC vaccines have been shown to be effective at inducing antigen-specific immune responses.57 Nanovaccines targeting STING have also been developed that potentiate antitumor T-cell activation.58,59 DCs pulsed with such vaccines and added to the donor graft may be effective as a novel cellular therapy to promote effective antitumor responses against minimal residual disease after HSCT. Some DC subsets have been identified as strong responders to STING-targeted agents and therefore could be isolated and/or expanded for adoptive cellular therapy accompanying HSCT.60 T1IFN has potent adjuvant activity in combinatorial approaches using DNA vaccines that target immature DCs.61 Interestingly, adjuvant activity of STING-targeted agents may be dependent on tumor necrosis factor α,62 an important cytokine produced after HSCT conditioning and during GVHD.63 Because STING has been shown to be a potent target for enhancing antitumor immunity, there is provocative rationale for developing cellular therapy approaches that use STING-targeted agents as adjuvants to eradicate residual tumor after HSCT.
Future directions and concluding remarks
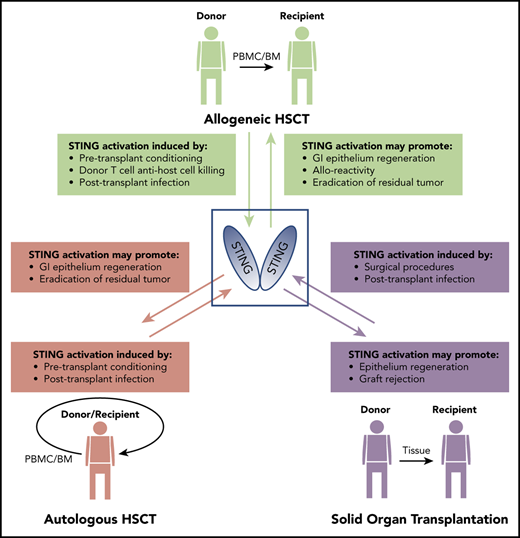
There is much diversity in the types of conditioning regimens used throughout various centers that perform allo-HSCT.64 We anticipate that, as the regimens involving radiation and/or chemotherapy (busulfan, fludarabine, and cyclophosphamide) are lessened, correspondingly less DNA damage will result which would decrease potential self-DNA–induced STING activation. In addition, reduced intensity regimens will result in diminished GI damage by decreasing leakage of bacterial products that activate STING.65 In contrast, high-dose myeloablative conditioning is the predominant choice for auto-HSCT conditioning.66,67 Because of cell death and accompanying GI damage resulting from myeloablative conditioning, STING signaling is likely pertinent in auto-HSCT and, lacking alloreactive T cells, would result from effects on host HCs and NH cells (Figure 1), including tumor cells. However, STING regulation at the time of auto-HSCT may suppress inflammation and epithelial repair, both potentially influencing nonrelapse mortality. STING agonists might also be used in combination with tumor vaccination to promote GVL in the auto-HSCT setting.68
Although it has not been explored, activation of the STING pathway would not be surprising after SOT. Cell death or DNA damage after SOT is likely less than that after HSCT but could provide a dsDNA source to activate STING as could bacterial CDNs after prolonged immunosuppression. Ischemia or reperfusion injury occurring during graft harvesting, cold storage, or surgery are underlying causes of inflammation and graft dysfunction.69-72 Subsequent activation of STING in HCs as well as NH-APCs in the tissue graft or draining nodes could drive host T-cell antidonor alloantigen rejection responses. Chronic rejection remains the most problematic challenge confronting transplantation physicians. Infection resulting from long-term immunosuppression provides potential for bacterial STING stimulation. The involvement of STING in the fate of solid tissue grafts could differ, depending on the location and type of transplanted tissue. For example, orthotopic ocular, skin, and GI allografts provide an environment that may readily expose the transplanted tissue cells to bacterial signals activating the STING pathway. In addition, use of immune suppressive treatment to prolong graft survival could, over time, lead to activation of host T-effector or memory cell populations that take up residence in the graft by persisting donor NH-APCs. It will be interesting to determine whether STING contributes via HCs and/or NH-APCs to SOT rejection and, if so, whether its targeting may be useful for prolonging graft survival.
aGVHD and cGVHD represent 2 distinct clinical diseases and have been demarcated by onset <100 and >100 days from allo-HSCT, respectively. However, it is well appreciated that aGVHD may not predate cGVHD and late-onset (>100 days) GVHD can be indistinguishable from aGVHD.73,74 Nonetheless, transition from aGVHD to cGVHD occurs and remains unexplained. Patients with defective immune systems who become exposed to infectious agents could experience sporadic or sustained STING activation weeks or months after HSCT that provokes alloantigen or autoantigen responses contributing to cGVHD.
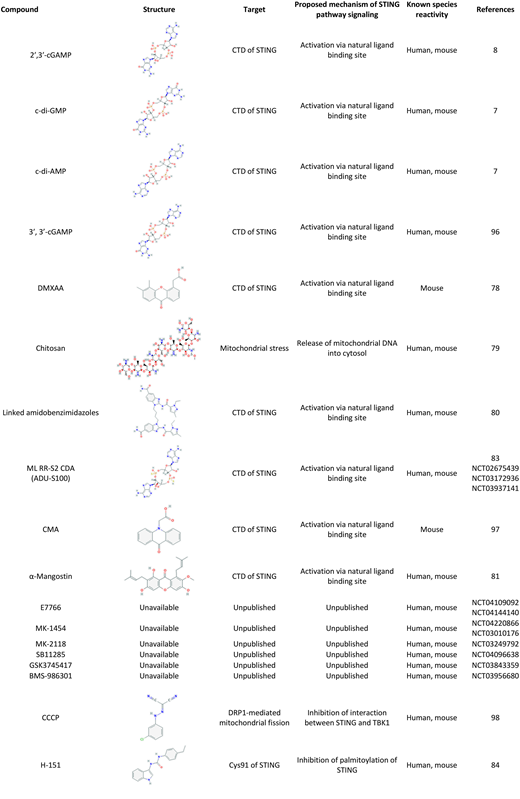
Preclinical studies in mice have taken advantage of gene deletion to generate STING-deficient animals that have enabled significant discoveries to improve understanding of the signaling pathway and its importance in host defenses. Interestingly, human STING alleles are heterogeneous, and R71H-G230A-R293Q (HAQ), the second most common STING allele, is associated with decreased STING function.11,75,76 STING can influence vaccine effectiveness because lower antibody responses were reported in STING-deficient mice.77 Our recent pneumococcal vaccine investigation identified diminished antibody responses in individuals with a single HAQ copy.75 With regard to targeting the STING pathway, although the initial compound targeting the ligand-binding region, 5,6-dimethylxanthenone-4-acetic acid (DMXAA), is ineffective in humans,78 chitosan, α-mangostin, and amidobenzimidazole-based compounds have been identified that agonize the STING pathway in humans and mice (Table 1).79-81 Studies investigating specific CDN family members have identified c-di-GMP and the novel synthetic CDN ML-RR-S2 c-di-AMP as potent STING activators.82,83 Nitrofuran-based small molecules can also inhibit human and mouse STING by targeting its transmembrane region (Table 1).84 Regarding clinical translation of mouse studies, STING’s broad expression and intracellular location will likely require adoptive cell therapy and/or targeting agonists and antagonists across the cell membrane to specific cell populations. For example, activating the STING pathway in APCs ex vivo before adoptive transfer to drive immune responses and targeting cells using nanoparticles directed to specific cell surface receptors that contain STING activators have been reported.85,86 These types of strategies would also diminish global and off-target effects.
STING agonists have also been successful as adjuvants for tumor vaccines.83,87 Regarding enhancement of GVL, delivery of STING agonists via nanoshells before chemotherapy can promote antitumor activity by placing tumor antigen and STING-based adjuvant in the same physical location.88 Liposomal nanoparticles loaded with CDNs are more effective at activating STING within tumor cells compared with naked CDNs, likely because of the impermeability of CDNs to the cell membrane.89 Combinatorial approaches that activate STING may be synergistic or additive with inhibitors of the noncanonical NF-κB, absent in melanoma 2 (AIM2) inflammasome, or caspase 1–mediated cell death pathways.90,91 STING activation via microbiota or agonists could also improve antitumor responses when combined with CD47, glucocorticoid-induced tumor necrosis factor receptor-related protein (GITR), or programmed cell death protein 1 (PD-1) checkpoint blockade.92,93 Murine studies have shown promising results for STING-targeting cancer therapeutics, and multiple trials implementing STING targeting for cancer immunotherapy of solid tumors are underway (E7766: NCT04109092, ADU-S100: NCT02675439, MK1454: NCT03010176) (Table 1). These agonists and others have not yet been tested for treatment of blood cancers and the promotion of the GVL effect. The use of posttransplant cyclophosphamide has been shown to diminish antirecipient alloreactivity.94,95 Administration of a tumor vaccine combined with STING agonists in patients at some time point after posttransplant cyclophosphamide could promote an antitumor-specific response without exacerbating GVHD alloreactivity.
In conclusion, isolating the absence of STING both singularly and in combination with donor cells, host HCs, host NH cells, or tumor cells will provide a way to carefully interrogate its significance in different tissues after experimental transplantation. Studies using STING pathway–specific agonists or antagonists could then be performed to corroborate the precise significance of the pathway in vivo and further define the location and sources of STING signals that influence the outcome of liquid and solid transplants.
Acknowledgments
The authors thank Zhibin Chen, Lazaros Lekakis, Sabrina Copsel, and Dietlinde Wolf for their critiques and helpful discussions regarding the manuscript.
This work was supported by grants from the National Institutes of Health (NIH), National Eye Institute (R01EY024484, R01EY030283), the Sylvester Comprehensive Cancer Center (R.B.L.), the NIH National Institute of Allergy and Infectious Diseases (R01AI110606, R21AI125999) (L.J.), and the NIH National Cancer Institute (F31CA216999, F99CA245728) (C.S.B.).
Authorship
Contribution: C.S.B., L.J., and R.B.L. wrote the manuscript.
Conflict-of-interest disclosure: R.B.L. served as a consultant or advisory board member for and equity holder in Heat Biologics. The remaining authors declare no competing financial interests.
Correspondence: Robert B. Levy, Department of Microbiology and Immunology, University of Miami Miller School of Medicine, R-138, 1600 NW 10th Ave, Miami, FL 33136; e-mail: rlevy@med.miami.edu.