In this issue of Blood, identify a mechanism that promotes cellular iron overload in Friedreich’s ataxia (FRDA), which is a result of a defect that prevents appropriate palmitoylation of transferrin receptor 1 (TfR1).1

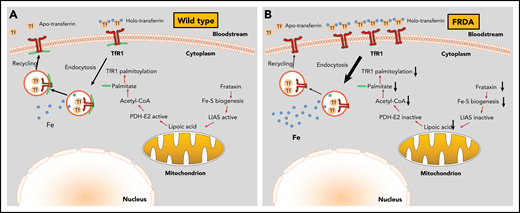
Mechanism for cellular iron overload in FRDA. (A) In wild-type cells, the presence of intact Fe-S assembly machinery allows proper TfR1 palmitoylation, plasma membrane expression, and iron uptake function. (B) In FRDA cells, frataxin deficiency impairs Fe-S biogenesis and inactivates LIAS, inhibiting lipoic acid synthesis. Because lipoic acid is an essential cofactor of the pyruvate dehydrogenase E2 subunit (PDH-E2), these responses limit acetyl-CoA production by PDH and subsequent fatty acid biosynthesis. In the absence of sufficient palmitate, TfR1 fails to undergo palmitoylation and accumulates on the plasma membrane. Moreover, the lack of TfR1 palmitoylation increases endocytosis of transferrin-TfR1 complexes and delays recycling of transferrin, which results in iron overload.
Mechanism for cellular iron overload in FRDA. (A) In wild-type cells, the presence of intact Fe-S assembly machinery allows proper TfR1 palmitoylation, plasma membrane expression, and iron uptake function. (B) In FRDA cells, frataxin deficiency impairs Fe-S biogenesis and inactivates LIAS, inhibiting lipoic acid synthesis. Because lipoic acid is an essential cofactor of the pyruvate dehydrogenase E2 subunit (PDH-E2), these responses limit acetyl-CoA production by PDH and subsequent fatty acid biosynthesis. In the absence of sufficient palmitate, TfR1 fails to undergo palmitoylation and accumulates on the plasma membrane. Moreover, the lack of TfR1 palmitoylation increases endocytosis of transferrin-TfR1 complexes and delays recycling of transferrin, which results in iron overload.
FRDA is an autosomal recessive neurodegenerative disease that is mainly caused by expansions of a GAA trinucleotide repeat within the first intron of the FXN gene.2 This impairs expression of the gene product frataxin, a mitochondrial protein involved in iron-sulfur (Fe-S) cluster (ISC) biogenesis. Frataxin deficiency is associated with mitochondrial iron overload, oxidative stress, and inactivation of ISC proteins such as mitochondrial aconitase and respiratory chain complexes I to III. These biochemical manifestations primarily affect the central nervous system and the heart and lead to sensory neuron dysfunction and cardiomyopathy.
The impact of frataxin deficiency on overall cellular iron metabolism is not well understood. Moreover, assessment of cytosolic iron status in cells and tissues from FRDA patients and mouse models has yielded conflicting results. It is generally believed that accumulation of mitochondrial iron is the result of an increased metabolic need for the metal because iron has not been incorporated into ISCs. This in turn drives iron flux from the cytosol into mitochondria. The ensuing cytosolic iron depletion is sensed by iron regulatory proteins IRP1 and IRP2, which stabilize the TfR1-encoding TFRC messenger RNA (mRNA) to increase iron uptake in a vicious cycle. At the same time, IRPs inhibit translation of the FTH, FTL, and SLC40A1 mRNAs that encode H-ferritin, L-ferritin, and ferroportin, respectively, to inhibit iron storage and export.3 Of note, proper regulation of IRPs requires functional ISC assembly machinery. Thus, in iron-replete cells, IRP1 acquires a 4Fe-4S cluster and operates as cytosolic aconitase at the expense of its RNA-binding activity. Conversely, IRP2 undergoes proteasomal degradation after ubiquitination by the E3 ubiquitin ligase FBXL5, which was recently shown to act by using a 2Fe-2S cluster.4 There is evidence that frataxin deficiency triggers activation of both IRP1 and IRP2 for RNA binding,5 which is consistent with cytosolic iron deficiency and defective ISC biogenesis.
Petit et al used skin fibroblasts from FRDA patients and showed that these cells have a higher iron content compared with controls in both cytosolic and mitochondrial compartments. Moreover, they fail to mount a negative feedback homeostatic response to exogenous holotransferrin or ferric ammonium citrate, and they develop excessive iron overload. FRDA fibroblasts exhibited similar TFRC mRNA expression compared with controls, which can be attributed to increased IRP2 activity, in spite of relatively low IRP2 expression. Surprisingly, frataxin deficiency did not promote induction of IRP1 for RNA binding, indicating an appropriate response to high iron content in spite of defective ISC assembly machinery. Another unexpected observation was that exogenous iron administration suppressed TFRC mRNA not only in controls but also in FRDA fibroblasts. However, the decrease in IRP2 expression was modest compared with the degree of iron accumulation. Together, these findings suggest a partial preservation of an IRP2-driven homeostatic response, which does not fully explain the dramatic susceptibility of FRDA fibroblasts to iron overload.
Petit et al noted that TfR1 steady-state levels were increased in iron-loaded FRDA fibroblasts notwithstanding the low TFRC mRNA expression. This prompted them to quantify the amount of TfR1 on the plasma membrane and compare it to the amount on control cells. Furthermore, they used live imaging via spinning disc microscopy to monitor endocytosis of transferrin-TfR1 complexes. These experiments identified a higher fraction of TfR1 present on the surface of FRDA fibroblasts, increased endocytosis, and a significant delay in the recycling of transferrin.
Earlier work established that this biochemical phenotype is linked to defective palmitoylation of TfR1.6 Palmitoylation is the covalent binding of palmitic acid via S-acyl radicals to cysteine residues of membrane proteins. Using an acyl-biotin exchange assay, Petit et al provided strong evidence for poor TfR1 palmitoylation in FRDA fibroblasts. In addition, they managed to decrease iron levels in these cells by using artesunate, an anti-malaria drug previously reported to stimulate TfR1 palmitoylation.7 In fact, the pharmacologic rescue of FRDA fibroblasts from iron overload was accompanied by proper TfR1 palmitoylation.
How is frataxin deficiency linked to defective TfR1 palmitoylation? Petit et al demonstrated that the disruption of ISC biogenesis in the absence of frataxin has a profound effect on intermediary metabolism by inhibiting generation of acetyl-coenzyme A (acetyl-CoA), the building block of fatty acids including palmitate. Dihydrolipoamide acetyltransferase (PDH-E2), a subunit of pyruvate dehydrogenase (PDH) that gives rise to acetyl-CoA, requires lipoic acid as a cofactor. This is produced by lipoic acid synthase (LIAS), an ISC enzyme that is inactive in FRDA fibroblasts. Importantly, supplementation of the cells with acetyl-CoA or pharmacologic stimulation of PDH activity by dichloroacetate restored PDH lipoylation and TfR1 palmitoylation. Moreover, targeted mitochondrial expression of exogenous frataxin decreased membrane TfR1 levels and corrected iron overload in skin fibroblasts, while artesunate also corrected iron overload in peripheral blood mononuclear cells from FRDA patients.
These data uncover a new mechanism for cellular iron overload in frataxin-deficient cells (see figure). Thus, the loss of frataxin inactivates Fe-S proteins, including LIAS, and thereby abrogates the synthesis of lipoic acid, an essential cofactor for acetyl-CoA production by PDH. Acetyl-CoA insufficiency limits fatty acid biosynthesis and TfR1 palmitoylation, which results in uncontrolled accumulation of TfR1 on the plasma membrane, increased iron transport activity, and eventually iron overload. Petit et al also identified reduced palmitoylation of the inorganic iron transporters DMT1 and ZIP14 in FRDA fibroblasts and were able to ameliorate iron overload imposed by ferric ammonium citrate using acetyl-CoA or artesunate. Conceivably, DMT1 and ZIP14 may contribute to iron overload in FRDA by internalizing non-transferrin-bound iron, but further validation of this hypothesis is required.
On a final note, the work of Petit et al raises the possibility of pharmacologic application of agents that restore acetyl-CoA production and TfR1 palmitoylation for managing patients with FRDA. It will be important to corroborate key findings in cells that are relevant to FRDA pathologies, such as neurons and cardiomyocytes, and also in animal models of FRDA.
Conflict-of-interest disclosure: The author declares no competing financial interests.