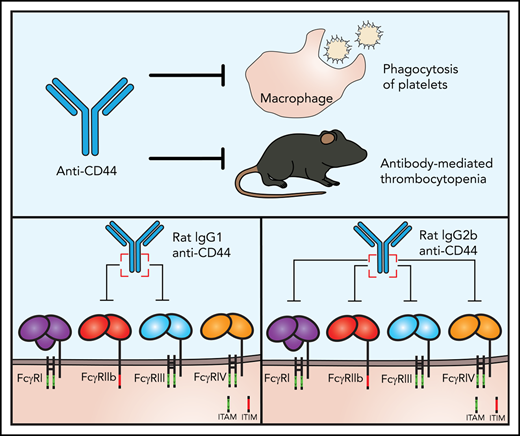
Key Points
CD44 antibodies block FcγR IgG binding function on macrophages.
CD44 antibody blockade of FcγRs leads to inhibition of macrophage phagocytosis of platelets and explains murine ITP amelioration.

Abstract
Monoclonal immunoglobulin G (IgG) antibodies to CD44 (anti-CD44) are anti-inflammatory in numerous murine autoimmune models, but the mechanisms are poorly understood. Anti-CD44 anti-inflammatory activity shows complete therapeutic concordance with IV immunoglobulin (IVIg) in treating autoimmune disease models, making anti-CD44 a potential IVIg alternative. In murine immune thrombocytopenia (ITP), there is no mechanistic explanation for anti-CD44 activity, although anti-CD44 ameliorates disease similarly to IVIg. Here, we demonstrate a novel anti-inflammatory mechanism of anti-CD44 that explains disease amelioration by anti-CD44 in murine ITP. Macrophages treated with anti-CD44 in vitro had dramatically suppressed phagocytosis through FcγRs in 2 separate systems of IgG-opsonized platelets and erythrocytes. Phagocytosis inhibition by anti-CD44 was mediated by blockade of the FcγR IgG binding site without changing surface FcγR expression. Anti-CD44 of different subclasses revealed that FcγR blockade was specific to receptors that could be engaged by the respective anti-CD44 subclass, and Fc-deactivated anti-CD44 variants lost all FcγR-inhibiting activity. In vivo, anti-CD44 functioned analogously in the murine passive ITP model and protected mice from ITP when thrombocytopenia was induced through an FcγR that could be engaged by the CD44 antibody’s subclass. Consistent with FcγR blockade, Fc-deactivated variants of anti-CD44 were completely unable to ameliorate ITP. Together, anti-CD44 inhibits macrophage FcγR function and ameliorates ITP consistent with an FcγR blockade mechanism. Anti-CD44 is a potential IVIg alternative and may be of particular benefit in ITP because of the significant role that FcγRs play in human ITP pathophysiology.
Introduction
CD44 is a broadly expressed receptor for hyaluronic acid and has been implicated in the disease processes of multiple autoimmune and inflammatory conditions. Targeting CD44 with monoclonal antibodies (anti-CD44) mediates significant anti-inflammatory effects in murine models of arthritis,1-6 chronic-relapsing experimental autoimmune encephalitis (EAE),7 experimental autoimmune uveoretinitis,8 and passive antibody-mediated immune thrombocytopenia (ITP).9,10 Anti-CD44 also shares essentially complete therapeutic concordance with IV immunoglobulin (IVIg) in treating murine autoimmune and inflammatory conditions, and it ameliorates murine ITP to a similar extent as IVIg but at a 3-log-fold lower dose.10 As such, we have proposed anti-CD44 as a potential IVIg alternative10 ; however, without a mechanism of action, transition to patient studies is impeded.
Primary ITP is an autoimmune condition with accelerated platelet clearance that is primarily due to anti-platelet autoantibodies.11,12 Autoantibodies against GPIIb/IIIa are the most prevalent in ITP and mediate platelet clearance largely through Fcγ receptors (FcγRs) on macrophages in the splenic mononuclear phagocyte system.13 This specific disease process is modeled by anti-GPIIb/IIIa antibodies in passive murine ITP14 ; like in humans, interfering with FcγR function is an effective therapeutic strategy in mice.14-17
Anti-CD44 demonstrates broad anti-inflammatory activity, including in murine ITP. However, the anti-inflammatory mechanism of anti-CD44 in murine ITP is not known. Given that the pathophysiology of murine passive ITP is antibody driven18 and antibodies play a significant role in other anti-CD44–treatable autoimmune models,19-21 we hypothesized that anti-CD44 is interfering with the ability of antibodies to mediate disease in ITP: specifically, that anti-CD44 engages with CD44 on phagocytes, leading to inhibition of phagocytosis and prevention of antibody-mediated platelet clearance.
Here, we report that the anti-inflammatory activity of anti-CD44 in murine ITP is explained by inhibition of the FcγR pathway. Using 2 monoclonal antibodies to CD44 for different immunoglobulin G (IgG) subclasses, we investigated the interaction between anti-CD44 and macrophage FcγRs and further translated these findings into the murine passive antibody-mediated ITP model. Anti-CD44 dramatically inhibited macrophage FcγR-mediated phagocytosis in 2 in vitro cytopenia models involving antibody-opsonized platelets and red blood cells (RBCs). Phagocytosis inhibition was dependent on the anti-CD44 Fc region and specifically targeted the FcγR IgG binding site. A proposed model for anti-CD44–mediated inhibition of macrophage FcγRs is given in Figure 1. Further, anti-CD44 functioned in complete concordance with this mechanism in vivo in the passive ITP model.
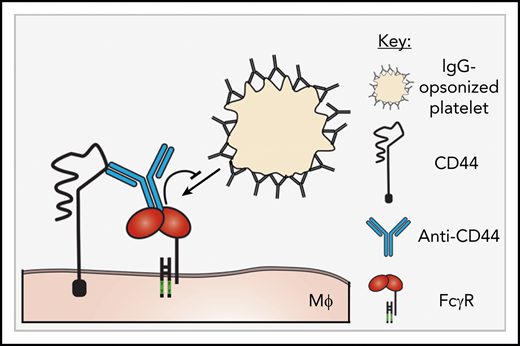
Proposed mechanism of anti-CD44-mediated FcγR inhibition. A proposed mechanism for anti-CD44–mediated inhibition of macrophage FcγRs. Anti-CD44’s antigen-binding region engages with CD44, and the Fc region of anti-CD44 binds to adjacent FcγRs, mediating blockade/inhibition of the FcγRs. Objects are not to scale. MΦ, macrophage.
Proposed mechanism of anti-CD44-mediated FcγR inhibition. A proposed mechanism for anti-CD44–mediated inhibition of macrophage FcγRs. Anti-CD44’s antigen-binding region engages with CD44, and the Fc region of anti-CD44 binds to adjacent FcγRs, mediating blockade/inhibition of the FcγRs. Objects are not to scale. MΦ, macrophage.
Methods
For full details, see supplemental Materials and methods, available on the Blood Web site.
Antibodies, mice, and cell culture
All antibodies were obtained from commercial suppliers with the exception of anti-GPIIb/IIIa clones 10B5 (mouse IgG2b), 2G1 (mouse IgG2a), and 5C4 (mouse IgG2a), which were generated in-house (as described in supplemental Materials and methods) and TER-119 variants with the same antigen binding and epitope specificity but switched to mouse IgG1 and mouse IgG2a, which were generated as previously described.22 IgG subclasses of in-house antibodies were confirmed by subclass-specific secondary antibody binding and flow cytometry, and clone 10B5 was additionally confirmed as IgG2b by liquid chromatography–tandem mass spectrometry (performed at Sparc BioCentre, The Hospital for Sick Children, Toronto, ON, Canada). C57BL/6 female mice and CD-1 female mice were purchased from Charles River Laboratories. FcR-γ chain–knockout mice [C.129P2(B6)-Fcer1gtm1RavN12]23 were purchased from Taconic Biosciences. FcγRIII-knockout mice (B6.129P2-Fcgr3tm1Jsv/J)24 were purchased from The Jackson Laboratory. RAW 264.7 macrophages were maintained and incubated at 37°C, 5% CO2 in complete RPMI, defined as RPMI pH 7.4 (MilliporeSigma Canada) supplemented with 10% fetal bovine serum (Wisent Bioproducts), antibiotic-antimycotic, and l-glutamine (both from Thermo Fisher Scientific).
Antibody deglycosylation and F(ab′)2 fragmentation
IgG1 anti-CD44 (KM114) and IgG2b anti-CD44 (IM7) were deglycosylated using glycerol-free recombinant PNGase-F (New England Biolabs). IgG2b anti-CD44 F(ab′)2 was prepared using Staphylococcus aureus V8 protease, as previously described.25 IgG1 anti-CD44 F(ab′)2 was prepared using ficin from a Pierce Mouse IgG1 Fab and F(ab′)2 Preparation Kit (Thermo Fisher Scientific), according to the manufacturer’s instructions, with slight modifications as described in supplemental Materials and methods.
Macrophage phagocytosis of antibody-opsonized RBCs
Macrophages (RAW 264.7) were seeded into wells of a 12-well polystyrene plate (Corning Inc.) that was placed in an incubator overnight (37°C, 5% CO2). Whole blood was collected from CD-1 mice and anticoagulated in a citrate solution. RBCs were obtained by centrifugation, and concentration was determined using a Guava easyCyte Flow Cytometer (Luminex Corp.). RBCs were opsonized with antibody for 30 minutes at room temperature. Concurrently, macrophages were treated with CD44 antibodies or controls for 30 minutes. Opsonized RBCs were added to macrophages at a 20:1 ratio, and phagocytosis proceeded for 30 minutes at 37°C, 5% CO2. Phagocytosis was stopped on ice, and nonphagocytosed RBCs were removed by hypotonic water lysis and phosphate-buffered saline (PBS; Sigma-Aldrich) washing before formaldehyde fixation. PBS washing was also performed between each macrophage manipulation and after RBC opsonization. Macrophages were imaged at the center of wells using a Nikon Eclipse TS100 inverted microscope. At least 4 images were taken per well (>500 cells counted).
Macrophage phagocytosis of antibody-opsonized platelets
Macrophages (RAW 264.7) were seeded on sterile glass coverslips (Thermo Fisher Scientific) inside wells of a 12-well polystyrene plate and incubated overnight (37°C, 5% CO2). Whole blood was collected from CD-1 mice into a citrate phosphate dextrose adenine buffer (MilliporeSigma) with carbacyclin (Santa Cruz Biotechnology) to inhibit platelet activation. Platelet-rich plasma was collected by centrifugation. Platelets were fluorescently labeled with 5-chloromethylfuorescein diacetate (Thermo Fisher Scientific) and then opsonized with anti-GPIIb/IIIa antibodies. Concurrently, macrophages were treated with anti-CD44 antibodies or controls for 30 minutes. Antibody-opsonized platelets were added to macrophages at a 100:1 ratio in complete RPMI. Phagocytosis was allowed to proceed for 30 minutes before stoppage on ice and formaldehyde fixation. Surface-bound platelets were detected using an anti-GPV antibody followed by Alexa Fluor 647–conjugated secondary antibody, incubated in sequence for 20 minutes each at room temperature. PBS washing was performed between each macrophage manipulation and each platelet step after collection. Coverslips were mounted on glass slides, and macrophages were observed by spinning-disc confocal microscopy. Images were reconstructed in 3 dimensions for analysis using Imaris v8.0.2 (Bitplane). Successfully phagocytosed platelets were visible by green fluorescence within macrophages and were distinguished from surface-bound and nonphagocytosed platelets by the absence of Alexa Fluor 647 staining.
Murine passive antibody-mediated ITP
ITP was passively induced by antibodies in C57BL/6 mice, as previously described.10 In brief, mice were administered 50 μg of IgG1 anti-CD44 (KM114), controls, or equimolar quantities of F(ab′)2 or deglycosylated variants by IV (tail vein) injection 30 minutes prior to IV injection of the anti-GPIIb/IIIa antibody (3 μg of clone MWReg30; 1 μg of clone 5C4). Platelet count was determined at 24 hours post–anti-GPIIb/IIIa injection using a Z2 Coulter Counter.
Flow cytometric analysis of FcγR expression and blockade
Macrophages were seeded into wells of a 12-well polystyrene plate in complete RPMI and placed in a CO2 incubator overnight. Macrophages were treated with anti-CD44, as indicated, or with the respective isotype controls for 30 minutes at 37°C, 5% CO2. Plates were then immediately placed on ice, and wells were washed with ice-cold PBS. Antibody-treated macrophages were fixed with formaldehyde (4% solution in PBS) on ice and washed repeatedly with PBS before incubation with fluorescent anti-FcγR antibodies. Macrophages were washed with PBS and detached by scraping, and flow cytometry was performed using a BD LSRFortessa X-20 (Becton Dickinson).
Data analysis
All statistical calculations were performed using GraphPad Prism version 7.04. Figures were prepared using GraphPad Prism version 7.04, Adobe Illustrator CC 2020 (Adobe Inc.), and Microsoft PowerPoint 2016. Flow cytometry data were analyzed using FlowJo version 10 (Becton Dickinson) and Kaluza Analysis version 2.1 (Beckman Coulter). Microscopy images were analyzed using Imaris version 8.0.2 (Bitplane) and Fiji (ImageJ).
Results
Anti-CD44 inhibits macrophage FcγR-mediated phagocytosis of platelets
Phagocyte FcγRs play an essential role in the clearance of platelets in the murine passive ITP model, particularly for antibodies against GPIIb/IIIa.14,26 Therefore, we investigated whether anti-CD44 can interfere with FcγR-mediated phagocytosis of anti-GPIIb/IIIa antibody–coated platelets. Macrophages (RAW 264.7) were treated with IgG1 anti-CD44 (rat, clone KM114), IgG2b anti-CD44 (rat, clone IM7), the FcγRIIb/III-blocking antibody 2.4G2, or isotype controls. Platelets opsonized with the rat IgG1 anti-GPIIb/IIIa antibody clone MWReg30 were added to macrophages for phagocytosis, and phagocytosis was visualized by microscopy (Figure 2A). Phagocytosis proceeded in a strictly IgG-dependent manner (Figure 2B). Treatment of macrophages with IgG1 or IgG2b anti-CD44 dramatically inhibited phagocytosis, reducing platelet uptake by >90% (Figure 2C). Phagocytosis of platelets was also blocked significantly by the FcγRIIb/III-blocking antibody 2.4G2, demonstrating that macrophage phagocytosis of rat IgG1 anti-GPIIb/IIIa–opsonized platelets required FcγRIII.
![Anti-CD44 inhibits macrophage FcγR-mediated phagocytosis of anti-GPIIb/IIIa–opsonized platelets. Macrophages (RAW 264.7) were treated with anti-CD44 or controls before incubation with platelets opsonized with anti-GPIIb/IIIa antibodies. (A) Confocal microscopy images of untreated macrophages (MΦ) (left panels) or macrophages treated with anti-CD44 (right panels) before incubation with anti-GPIIb/IIIa (clone MWReg30)–opsonized platelets. Scale bars, 10 μm. All platelets (internalized and external) are labeled with the green cytoplasmic dye 5-chloromethylfuorescein diacetate (upper panels). External nonphagocytosed platelets were distinguished after completion of phagocytosis and formaldehyde fixation with an anti–glycoprotein V (GPV) primary antibody plus Alexa Fluor 647 (AF-647) secondary antibody (lower panels). Arrowheads point to example macrophages with phagocytosed platelets. Images were taken using a Quorum multimodal imaging system with a 63× oil-immersion objective (NA 1.47). Fluorescent images are merged with differential interference contrast images. Images were Z-stacked every 330 nm and reconstructed in 3 dimensions for analysis using Imaris v8.0.2. At least 4 images were taken at the center of each well (>500 cells counted). (B) Magnitude of macrophage phagocytosis of anti-GPIIb/IIIa IgG1 (clone MWReg30)–opsonized platelets. Nonopsonized platelets (-) were incubated with PBS only during the opsonization step before addition to macrophages. The phagocytic index is the average number of phagocytosed platelets per 100 macrophages. Data are from 6 experiments (mean ± standard error of the mean [SEM]). **P = .0022, nonparametric Mann-Whitney t test. (C) Effect of anti-CD44 (α-CD44) antibodies of different IgG subclasses (rat IgG1 anti-CD44 clone KM114 and rat IgG2b anti-CD44 clone IM7) on macrophage phagocytosis of IgG1-opsonized platelets. FcγRIII block is antibody clone 2.4G2. Data (mean ± SEM) are from 4 or 5 experiments. Dotted line indicates the level of phagocytosis by untreated macrophages (100%). The percentage of phagocytosis was calculated as (PI treatment/PI untreated control) × 100%, where PI (phagocytic index) is the average number of phagocytosed platelets per 100 macrophages. ****P < .0001, 1-way analysis of variance with Tukey’s post hoc test. CMFDA, 5-chloromethylfuorescein diacetate.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/137/15/10.1182_blood.2020009497/1/m_bloodbld2020009497f2.png?Expires=1769309127&Signature=f7v8AZVthb5FeQhjNolBRxq8BRZlWY~OdhuGBXQKhErpNDeaoqwDv6g0WIM-5v3fxtxb72aRnYhRIKEYoL~6ZDBYYDd-ORLLXzEEIVIaa~bwlUsRfQv4qIWTe6-7qq6GTEPnmzwv2VAQPug1r0ck9BdgN0OawQffgYHHmpAKmdxY3qZQD83LvGhieXt2Uo3q0YLZ-wFDYyMDo7ElD95Bo3povo73rVHhrs3v5Cbi62GrMbI20HrT~Jw1zjstDUV9DNPRtwJ9ToTO48HloO2dCUYffFlRT3axCTYRG7vfBXgYaLO8G7ZU7L0JushcTe8xxcEoq3wxtfwo8og7blhgBw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Anti-CD44 inhibits macrophage FcγR-mediated phagocytosis of anti-GPIIb/IIIa–opsonized platelets. Macrophages (RAW 264.7) were treated with anti-CD44 or controls before incubation with platelets opsonized with anti-GPIIb/IIIa antibodies. (A) Confocal microscopy images of untreated macrophages (MΦ) (left panels) or macrophages treated with anti-CD44 (right panels) before incubation with anti-GPIIb/IIIa (clone MWReg30)–opsonized platelets. Scale bars, 10 μm. All platelets (internalized and external) are labeled with the green cytoplasmic dye 5-chloromethylfuorescein diacetate (upper panels). External nonphagocytosed platelets were distinguished after completion of phagocytosis and formaldehyde fixation with an anti–glycoprotein V (GPV) primary antibody plus Alexa Fluor 647 (AF-647) secondary antibody (lower panels). Arrowheads point to example macrophages with phagocytosed platelets. Images were taken using a Quorum multimodal imaging system with a 63× oil-immersion objective (NA 1.47). Fluorescent images are merged with differential interference contrast images. Images were Z-stacked every 330 nm and reconstructed in 3 dimensions for analysis using Imaris v8.0.2. At least 4 images were taken at the center of each well (>500 cells counted). (B) Magnitude of macrophage phagocytosis of anti-GPIIb/IIIa IgG1 (clone MWReg30)–opsonized platelets. Nonopsonized platelets (-) were incubated with PBS only during the opsonization step before addition to macrophages. The phagocytic index is the average number of phagocytosed platelets per 100 macrophages. Data are from 6 experiments (mean ± standard error of the mean [SEM]). **P = .0022, nonparametric Mann-Whitney t test. (C) Effect of anti-CD44 (α-CD44) antibodies of different IgG subclasses (rat IgG1 anti-CD44 clone KM114 and rat IgG2b anti-CD44 clone IM7) on macrophage phagocytosis of IgG1-opsonized platelets. FcγRIII block is antibody clone 2.4G2. Data (mean ± SEM) are from 4 or 5 experiments. Dotted line indicates the level of phagocytosis by untreated macrophages (100%). The percentage of phagocytosis was calculated as (PI treatment/PI untreated control) × 100%, where PI (phagocytic index) is the average number of phagocytosed platelets per 100 macrophages. ****P < .0001, 1-way analysis of variance with Tukey’s post hoc test. CMFDA, 5-chloromethylfuorescein diacetate.
Anti-CD44 inhibits macrophage FcγR-mediated phagocytosis of anti-GPIIb/IIIa–opsonized platelets. Macrophages (RAW 264.7) were treated with anti-CD44 or controls before incubation with platelets opsonized with anti-GPIIb/IIIa antibodies. (A) Confocal microscopy images of untreated macrophages (MΦ) (left panels) or macrophages treated with anti-CD44 (right panels) before incubation with anti-GPIIb/IIIa (clone MWReg30)–opsonized platelets. Scale bars, 10 μm. All platelets (internalized and external) are labeled with the green cytoplasmic dye 5-chloromethylfuorescein diacetate (upper panels). External nonphagocytosed platelets were distinguished after completion of phagocytosis and formaldehyde fixation with an anti–glycoprotein V (GPV) primary antibody plus Alexa Fluor 647 (AF-647) secondary antibody (lower panels). Arrowheads point to example macrophages with phagocytosed platelets. Images were taken using a Quorum multimodal imaging system with a 63× oil-immersion objective (NA 1.47). Fluorescent images are merged with differential interference contrast images. Images were Z-stacked every 330 nm and reconstructed in 3 dimensions for analysis using Imaris v8.0.2. At least 4 images were taken at the center of each well (>500 cells counted). (B) Magnitude of macrophage phagocytosis of anti-GPIIb/IIIa IgG1 (clone MWReg30)–opsonized platelets. Nonopsonized platelets (-) were incubated with PBS only during the opsonization step before addition to macrophages. The phagocytic index is the average number of phagocytosed platelets per 100 macrophages. Data are from 6 experiments (mean ± standard error of the mean [SEM]). **P = .0022, nonparametric Mann-Whitney t test. (C) Effect of anti-CD44 (α-CD44) antibodies of different IgG subclasses (rat IgG1 anti-CD44 clone KM114 and rat IgG2b anti-CD44 clone IM7) on macrophage phagocytosis of IgG1-opsonized platelets. FcγRIII block is antibody clone 2.4G2. Data (mean ± SEM) are from 4 or 5 experiments. Dotted line indicates the level of phagocytosis by untreated macrophages (100%). The percentage of phagocytosis was calculated as (PI treatment/PI untreated control) × 100%, where PI (phagocytic index) is the average number of phagocytosed platelets per 100 macrophages. ****P < .0001, 1-way analysis of variance with Tukey’s post hoc test. CMFDA, 5-chloromethylfuorescein diacetate.
Anti-CD44 inhibits distinct FcγR pathways according to IgG subclass
We explored the mechanism behind anti-CD44’s inhibition of FcγR-dependent phagocytosis by selecting 3 additional anti-GPIIb/IIIa antibodies to drive phagocytosis through different FcγRs: 2 murine IgG2a antibodies (clones 2G1 and 5C4) and murine IgG2b clone 10B5. The properties of these anti-GPIIb/IIIa antibodies are summarized in Table 1. Murine IgG1 antibodies interact with only 1 activating FcγR (FcγRIII) and, therefore, are FcγRIII dependent; this paradigm is also true for the rat IgG1 antibody MWReg30.14,27 In contrast, murine IgG2a antibodies are not solely dependent on FcγRIII for activity and can engage multiple activating FcγRs (FcγRI, FcγRIII, and FcγRIV)27 and, therefore, are FcγRIII nondependent. All GPIIb/IIIa antibodies mediated significant platelet uptake by macrophages (Figure 3A). Both murine IgG2a antibodies mediated FcγRIII-nondependent phagocytosis, as established using the FcγRIII-blocking antibody 2.4G2 (Figure 3B). In contrast, MWReg30 (rat IgG1) was completely FcγRIII dependent (Figure 3B). Clone 10B5 (murine IgG2b) was also FcγRIII dependent, although murine IgG2b is typically reported as being able to engage all FcγRs.27 Clone 10B5 was confirmed as murine IgG2b, κ by mass spectrometry (data not shown).
![Anti-CD44 IgG subclass determines the specific macrophage FcγRs inhibited. Macrophages were treated with anti-CD44 or controls before incubation with platelets opsonized with anti-GPIIb/IIIa antibodies of different IgG subclasses. (A) Magnitude of untreated macrophage phagocytosis of platelets opsonized with different anti-GPIIb/IIIa IgG subclasses. Nonopsonized platelets (-) were incubated with PBS only during the opsonization step before addition to macrophages. Data are from 6 to 11 experiments (mean ± standard error of the mean [SEM]). Effect of FcγRIII blockade (clone 2.4G2) (B), rat IgG1 anti-CD44 (C), or rat IgG2b anti-CD44 (D) compared with no antibody treatment (-) on macrophage phagocytosis of platelets opsonized with the different IgG subclasses shown. Data are from 3 or 4 experiments (mean ± SEM). The phagocytic index is the average number of phagocytosed platelets per 100 macrophages. ****P < .0001, ***P < .001, **P < .01, vs the nonopsonized group, nonparametric Mann-Whitney t test (A), unpaired t test (B-D). IgG1, rat IgG1 clone MWReg30; IgG2a, mouse IgG2a (clone 5C4 or 2G1); IgG2b, mouse IgG2b clone 10B5; ns, not significant; α-CD44, anti-CD44.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/137/15/10.1182_blood.2020009497/1/m_bloodbld2020009497f3.png?Expires=1769309127&Signature=0yHsd-tNQ0bu8Af6EWa~6y3EOTjYFXDXXGY9G8~tA4n3IRSi6SdP5BQU269RJabKaH-iU6p9Eo6oTqejIRfNTPquifhjURD08MUpGTfKLjKc38D1Jk9IYs~Ih4vDGdrO2zoOBWKU9c-4GGtWUfOqnGk~f5v9fDCYw17WOggOSs8lruwvL6CBLgT35VF1tO5Y1LtGlKljjaeZH1mw79yUxERFciRlT00P9W-0CVqkfmZ4qo3g5TChDbzgQsWAPDgdyMhTPuQ1lX9yzhMixYTD3vMALBPVopxzNC6NiNIs0m0ylAlGMAYN0~zStArcmbGSt1KhlUiFCtaSH6elooC9cg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Anti-CD44 IgG subclass determines the specific macrophage FcγRs inhibited. Macrophages were treated with anti-CD44 or controls before incubation with platelets opsonized with anti-GPIIb/IIIa antibodies of different IgG subclasses. (A) Magnitude of untreated macrophage phagocytosis of platelets opsonized with different anti-GPIIb/IIIa IgG subclasses. Nonopsonized platelets (-) were incubated with PBS only during the opsonization step before addition to macrophages. Data are from 6 to 11 experiments (mean ± standard error of the mean [SEM]). Effect of FcγRIII blockade (clone 2.4G2) (B), rat IgG1 anti-CD44 (C), or rat IgG2b anti-CD44 (D) compared with no antibody treatment (-) on macrophage phagocytosis of platelets opsonized with the different IgG subclasses shown. Data are from 3 or 4 experiments (mean ± SEM). The phagocytic index is the average number of phagocytosed platelets per 100 macrophages. ****P < .0001, ***P < .001, **P < .01, vs the nonopsonized group, nonparametric Mann-Whitney t test (A), unpaired t test (B-D). IgG1, rat IgG1 clone MWReg30; IgG2a, mouse IgG2a (clone 5C4 or 2G1); IgG2b, mouse IgG2b clone 10B5; ns, not significant; α-CD44, anti-CD44.
Anti-CD44 IgG subclass determines the specific macrophage FcγRs inhibited. Macrophages were treated with anti-CD44 or controls before incubation with platelets opsonized with anti-GPIIb/IIIa antibodies of different IgG subclasses. (A) Magnitude of untreated macrophage phagocytosis of platelets opsonized with different anti-GPIIb/IIIa IgG subclasses. Nonopsonized platelets (-) were incubated with PBS only during the opsonization step before addition to macrophages. Data are from 6 to 11 experiments (mean ± standard error of the mean [SEM]). Effect of FcγRIII blockade (clone 2.4G2) (B), rat IgG1 anti-CD44 (C), or rat IgG2b anti-CD44 (D) compared with no antibody treatment (-) on macrophage phagocytosis of platelets opsonized with the different IgG subclasses shown. Data are from 3 or 4 experiments (mean ± SEM). The phagocytic index is the average number of phagocytosed platelets per 100 macrophages. ****P < .0001, ***P < .001, **P < .01, vs the nonopsonized group, nonparametric Mann-Whitney t test (A), unpaired t test (B-D). IgG1, rat IgG1 clone MWReg30; IgG2a, mouse IgG2a (clone 5C4 or 2G1); IgG2b, mouse IgG2b clone 10B5; ns, not significant; α-CD44, anti-CD44.
Strikingly, anti-CD44 inhibited selective FcγR-mediated phagocytosis pathways according to CD44 antibody IgG subclass. IgG1 anti-CD44 inhibited phagocytosis of platelets opsonized with the FcγRIII-dependent subclasses (rat IgG1 clone MWReg30 and mouse IgG2b clone 10B5) but not with the FcγRIII-nondependent murine IgG2a antibodies (Figure 3C). In contrast, IgG2b anti-CD44 inhibited phagocytosis mediated by all anti-GPIIb/IIIa IgG subclasses, suggesting broad FcγR inhibition (Figure 3D).
The Fc region of anti-CD44 is required for inhibition of FcγR-mediated phagocytosis
Anti-CD44 antibodies of rat IgG1 and IgG2b subclasses were potent inhibitors of phagocytosis but appeared to impinge on different FcγR pathways. Because the specific FcγR pathways inhibited matched the activating FcγR binding profiles of the anti-CD44 IgG subclasses as rat IgG1 binds only FcγRIII,14 whereas rat IgG2b binds all activating FcγRs28 (confirmed in supplemental Figure 1), we hypothesized that the Fc region of anti-CD44 was involved. Therefore, we converted the IgG1 and IgG2b anti-CD44 antibodies into fully Fc-deglycosylated variants using PNGase-F (which substantially reduces FcγR binding) or F(ab′)2 fragments and assessed these variants for activity against macrophage phagocytosis. Substantial reduction and elimination of murine FcγR binding by deglycosylated and F(ab′)2 anti-CD44, respectively, were confirmed by enzyme-linked immunosorbent assay (supplemental Figure 1). The deglycosylated and F(ab′)2 variants of both anti-CD44 IgG subclasses were completely unable to inhibit macrophage phagocytosis of platelets for all opsonizing anti-platelet IgG subclasses tested (Figure 4), confirming an essential role for the CD44 antibody Fc region in FcγR inhibition.
![The Fc region of anti-CD44 is critically required to inhibit macrophage FcγR-mediated phagocytosis. Macrophages were treated with unmodified (intact) anti-CD44 or with Fc-deactivated variants [deglycosylated and F(ab′)2]. Platelets opsonized with different anti-GPIIb/IIIa IgG subclasses were added to macrophages to induce phagocytosis. (A-D) Macrophages were treated with unmodified anti-CD44 or Fc-deactivated anti-CD44 variants before incubation with platelets opsonized with different IgG subclasses. Dotted lines indicate the level of phagocytosis by untreated macrophages (100%). Data are from 3 to 5 experiments (mean ± standard error of the mean). The percentage of phagocytosis was calculated as (PI treatment/PI untreated control) × 100%, where PI (phagocytic index) is the average number of phagocytosed platelets per 100 macrophages. ****P < .0001, ***P < .001, 1-way analysis of variance with Tukey’s post hoc test. Degly., deglycosylated; IgG1, rat IgG1 (MWReg30); IgG2a, mouse IgG2a (2G1 or 5C4); IgG2b, mouse IgG2b (10B5).](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/137/15/10.1182_blood.2020009497/1/m_bloodbld2020009497f4.png?Expires=1769309127&Signature=O1x30rfZ~BnQsW2lMpqIwG4Qk4bHplXKQ8zbHfAoXKhGsRVEeb3469KONXOc6PT21-w5LFwgjt6eh5C4uN3rqlukhrzLDiwFI-aGpnEDOs7qnrcECq8HtoBhSeaW0q85H2mJSiAwOq7V6IrwKJzyZo8PGl1zM7WK6RlzXZkoYbT21-IV5atdzsaJ2eIDIaxq5OpH9R29jUIFuateuEwnHoJwDthi3KjE4oht5f5h8Eoktq4yfWeFNKlkwmKj~hMjdvegVHwzim-3QZSshXeFttsjmx8tCvudSJyyjraewFJzAPbCPiXMzr0zmDMcXFVRmKY67y55FdJYXGFj8TMp6w__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
The Fc region of anti-CD44 is critically required to inhibit macrophage FcγR-mediated phagocytosis. Macrophages were treated with unmodified (intact) anti-CD44 or with Fc-deactivated variants [deglycosylated and F(ab′)2]. Platelets opsonized with different anti-GPIIb/IIIa IgG subclasses were added to macrophages to induce phagocytosis. (A-D) Macrophages were treated with unmodified anti-CD44 or Fc-deactivated anti-CD44 variants before incubation with platelets opsonized with different IgG subclasses. Dotted lines indicate the level of phagocytosis by untreated macrophages (100%). Data are from 3 to 5 experiments (mean ± standard error of the mean). The percentage of phagocytosis was calculated as (PI treatment/PI untreated control) × 100%, where PI (phagocytic index) is the average number of phagocytosed platelets per 100 macrophages. ****P < .0001, ***P < .001, 1-way analysis of variance with Tukey’s post hoc test. Degly., deglycosylated; IgG1, rat IgG1 (MWReg30); IgG2a, mouse IgG2a (2G1 or 5C4); IgG2b, mouse IgG2b (10B5).
The Fc region of anti-CD44 is critically required to inhibit macrophage FcγR-mediated phagocytosis. Macrophages were treated with unmodified (intact) anti-CD44 or with Fc-deactivated variants [deglycosylated and F(ab′)2]. Platelets opsonized with different anti-GPIIb/IIIa IgG subclasses were added to macrophages to induce phagocytosis. (A-D) Macrophages were treated with unmodified anti-CD44 or Fc-deactivated anti-CD44 variants before incubation with platelets opsonized with different IgG subclasses. Dotted lines indicate the level of phagocytosis by untreated macrophages (100%). Data are from 3 to 5 experiments (mean ± standard error of the mean). The percentage of phagocytosis was calculated as (PI treatment/PI untreated control) × 100%, where PI (phagocytic index) is the average number of phagocytosed platelets per 100 macrophages. ****P < .0001, ***P < .001, 1-way analysis of variance with Tukey’s post hoc test. Degly., deglycosylated; IgG1, rat IgG1 (MWReg30); IgG2a, mouse IgG2a (2G1 or 5C4); IgG2b, mouse IgG2b (10B5).
Anti-CD44 inhibition of macrophage phagocytosis is not limited to platelets
To rule out whether the observed anti-CD44–mediated inhibition of phagocytosis is a platelet-specific phenomenon, IgG-opsonized murine RBCs were used as the phagocytic target instead. RBCs were opsonized with the anti-RBC antibody TER-119 (rat IgG2b) or TER-119 switched to mouse IgG1 and mouse IgG2a subclasses (supplemental Figure 2). The properties of these anti-RBC antibodies are provided in Table 1.
Similar to the platelet scenario, IgG1 anti-CD44 blocked macrophage phagocytosis of RBCs opsonized with FcγRIII-dependent antibodies (mouse IgG1-TER-119) but did not have any effect against FcγRIII-nondependent antibodies (mouse IgG2a and rat IgG2b TER-119) (supplemental Figure 2D). In contrast, IgG2b anti-CD44 broadly inhibited macrophage phagocytosis of RBCs opsonized with any TER-119 IgG subclass (supplemental Figure 2E). When modified to deglycosylated and F(ab′)2 variants, anti-CD44 of either subclass no longer inhibited phagocytosis [F(ab')2], or mediated substantially less inhibition (deglycosylated) (supplemental Figure 2F-H), supporting that anti-CD44 inhibition of phagocytosis is not specific to platelets and occurs with a separate IgG-opsonized target by a similar mechanism.
Anti-CD44 inhibits FcγRs by blocking the FcγR IgG binding site
We hypothesized that the anti-CD44 Fc region is binding and blocking the IgG binding site of FcγRs (Figure 1). To assess the effect of anti-CD44 on the FcγR IgG binding site vs change in FcγR expression, we evaluated the binding of blocking vs nonblocking anti-FcγR antibodies after anti-CD44 treatment. The use of nonblocking anti-FcγR antibodies allowed us to evaluate FcγR expression, even if the receptor may be occupied with IgG.
Clone 275003 was used as a blocking antibody, because it blocks binding of immune complexes to FcγRIII29 and blockades FcγRIII function in phagocytosis (supplemental Figure 3A). In contrast, clone 015 was used as a nonblocking antibody because it does not blockade FcγRIII function in phagocytosis (supplemental Figure 3A) and is not blocked by immune complexes for binding to FcγRs (supplemental Figure 3B).
Macrophages were treated with anti-CD44 (IgG1 or IgG2b) followed by fixation and incubation with the blocking antibody 275003 or nonblocking antibody 015. IgG1 and IgG2b CD44 antibodies dramatically blocked the binding of clone 275003, suggesting near-complete blockade of the FcγRIII IgG binding site (Figure 5A). In contrast, clone 015 binding was completely unchanged, indicating that anti-CD44 did not cause decreased FcγRIII surface expression (Figure 5B).

Anti-CD44 blocks the FcγR IgG binding site. The effect of anti-CD44 on macrophage FcγR expression and blockade of the FcγR IgG binding site was evaluated using blocking and nonblocking monoclonal antibodies to FcγRs. Macrophages were treated with anti-CD44 or controls for 30 minutes at 37°C, followed by washing, formaldehyde fixation, and addition of fluorescent anti-FcγR antibody. (A-B) Blocking and nonblocking anti-FcγRIII antibodies were used to distinguish FcγR blockade of the IgG binding site vs loss of FcγR expression. (A) Binding of a blocking FcγRIII-specific antibody (clone 275003) to macrophages that were treated or not (Nil) with IgG1 anti-CD44 (KM114) or IgG2b anti-CD44 (IM7). A representative line graph (left panel) and quantitation of 3 experiments (right panel) are shown. The gray shaded area (right panel) indicates the background fluorescence. Counts (%max) are counts expressed as a percentage relative to the mode (100%). (B) Binding of a nonblocking anti-FcγRIIb/III antibody (clone 015) to macrophages that were treated or not (Nil) with IgG1 anti-CD44 (KM114) or IgG2b anti-CD44 (IM7). A representative line graph (left panel) and quantitation of 3 experiments (right panel) are shown. The dotted line (right panel) indicates the background fluorescence. (C) The requirement of the CD44 antibody Fc region to block binding of the anti-FcγRIII antibody (275003 binding) was evaluated with unmodified anti-CD44 (Intact), anti-CD44 F(ab′)2 variants, or respective isotype controls (n = 3-8 experiments). The dotted line indicates the level of 275003 binding on untreated macrophages (100%). (D) Binding of an FcγRIV-blocking antibody (clone 9E9) after macrophage incubation with unmodified anti-CD44, anti-CD44 F(ab′)2 variants, or respective isotype controls (n = 3-5 experiments). The dotted line indicates the level of 9E9 binding on untreated macrophages (100%). The percentage of anti-FcγR antibody binding in (C) and (D) was calculated relative to fluorescent antibody MFI on untreated macrophages. Error: mean ± standard error of the mean. ****P < .0001, ***P < .001, *P < .05, 1-way analysis of variance with Tukey’s post hoc test. α-CD44, anti-CD44; MFI, mean fluorescence intensity.
Anti-CD44 blocks the FcγR IgG binding site. The effect of anti-CD44 on macrophage FcγR expression and blockade of the FcγR IgG binding site was evaluated using blocking and nonblocking monoclonal antibodies to FcγRs. Macrophages were treated with anti-CD44 or controls for 30 minutes at 37°C, followed by washing, formaldehyde fixation, and addition of fluorescent anti-FcγR antibody. (A-B) Blocking and nonblocking anti-FcγRIII antibodies were used to distinguish FcγR blockade of the IgG binding site vs loss of FcγR expression. (A) Binding of a blocking FcγRIII-specific antibody (clone 275003) to macrophages that were treated or not (Nil) with IgG1 anti-CD44 (KM114) or IgG2b anti-CD44 (IM7). A representative line graph (left panel) and quantitation of 3 experiments (right panel) are shown. The gray shaded area (right panel) indicates the background fluorescence. Counts (%max) are counts expressed as a percentage relative to the mode (100%). (B) Binding of a nonblocking anti-FcγRIIb/III antibody (clone 015) to macrophages that were treated or not (Nil) with IgG1 anti-CD44 (KM114) or IgG2b anti-CD44 (IM7). A representative line graph (left panel) and quantitation of 3 experiments (right panel) are shown. The dotted line (right panel) indicates the background fluorescence. (C) The requirement of the CD44 antibody Fc region to block binding of the anti-FcγRIII antibody (275003 binding) was evaluated with unmodified anti-CD44 (Intact), anti-CD44 F(ab′)2 variants, or respective isotype controls (n = 3-8 experiments). The dotted line indicates the level of 275003 binding on untreated macrophages (100%). (D) Binding of an FcγRIV-blocking antibody (clone 9E9) after macrophage incubation with unmodified anti-CD44, anti-CD44 F(ab′)2 variants, or respective isotype controls (n = 3-5 experiments). The dotted line indicates the level of 9E9 binding on untreated macrophages (100%). The percentage of anti-FcγR antibody binding in (C) and (D) was calculated relative to fluorescent antibody MFI on untreated macrophages. Error: mean ± standard error of the mean. ****P < .0001, ***P < .001, *P < .05, 1-way analysis of variance with Tukey’s post hoc test. α-CD44, anti-CD44; MFI, mean fluorescence intensity.
We further validated the effect of anti-CD44 on the FcγRIII IgG binding site using anti-CD44 F(ab′)2 variants. The reduction in 275003 binding after anti-CD44 was completely dependent on the anti-CD44 Fc region (Figure 5C), supporting that anti-CD44 blocks FcγRIII by using its Fc region. Blockade of FcγRIII did not appear to require interactions with neighboring macrophages, because decreasing the cell density to yield isolated macrophages did not change anti-CD44 blockade of FcγRIII (supplemental Figure 4). Anti-CD44 also blocked heat-aggregated IVIg immune complex binding to macrophages through an Fc-dependent mechanism (data not shown).
The effect of anti-CD44 on a separate FcγR (FcγRIV) was evaluated using the well-characterized FcγRIV blocking antibody 9E9.30 IgG2b anti-CD44 significantly reduced 9E9 binding, and this effect was dependent on the anti-CD44 Fc region (Figure 5D). In contrast, IgG1 anti-CD44 did not significantly block the binding of clone 9E9 to FcγRIV (Figure 5D), consistent with the FcγRIII-restricted nature of IgG1 antibodies.
Anti-CD44 ameliorates murine passive ITP in an FcγR-specific manner
CD44 antibodies share broad therapeutic concordance with IVIg and ameliorate murine ITP at a 3-log-fold lower dose.9,10 However, there is no mechanistic explanation for anti-CD44 activity in murine ITP. Therefore, we next evaluated whether the observed mechanism of anti-CD44 in vitro may explain anti-CD44 activity in vivo.
We first asked whether the IgG1 anti-CD44 antibody, which specifically blocked FcγRIII in vitro, also only ameliorates FcγRIII-mediated thrombocytopenia in the murine passive ITP model. Mice were injected IV with IgG1 anti-CD44 or an isotype control before induction of ITP with anti-GPIIb/IIIa antibodies MWReg30 (rat IgG1) or 5C4 (mouse IgG2a), and platelet counts were evaluated 24 hours later. MWReg30 was confirmed to mediate FcγRIII-dependent ITP, whereas 5C4 was confirmed to mediate ITP through an FcγRIII-nondependent, but FcRγ chain–dependent, manner (supplemental Figures 5 and 6).
Matching the FcγR-specific effects of anti-CD44 on phagocytosis observed in vitro, IgG1 anti-CD44 mediated significant protection against FcγRIII-mediated thrombocytopenia (Figure 6A) but was completely unable to prevent thrombocytopenia mediated through other FcγRs (Figure 6B).
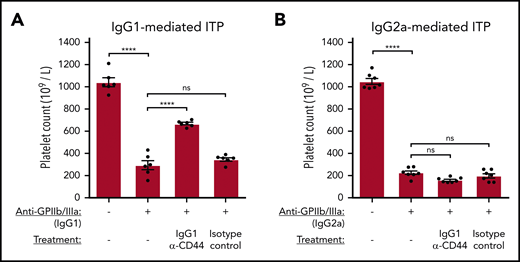
Anti-CD44 inhibits FcγR-mediated platelet clearance according to anti-GPIIb/IIIa IgG subclass in murine ITP. Passive antibody-mediated ITP was induced in C57BL/6 mice via IV tail vein injection using IgG1 anti-GPIIb/IIIa (clone MWReg30), which mediates thrombocytopenia through FcγRIII (A), or IgG2a anti-GPIIb/IIIa (clone 5C4), which mediates thrombocytopenia through FcγRs but is not dependent on FcγRIII (B). Anti-CD44 (α-CD44) or isotype control was administered IV by tail vein injection 30 minutes before anti-platelet antibody injection. Platelet counts were determined at 24 hours after anti-platelet antibody administration. Data are from 6 (A) or 7 (B) independent experiments with 1 mouse per experiment. Error: mean ± standard error of the mean (SEM). ****P < .0001, 1-way analysis of variance with Tukey’s post hoc test.
Anti-CD44 inhibits FcγR-mediated platelet clearance according to anti-GPIIb/IIIa IgG subclass in murine ITP. Passive antibody-mediated ITP was induced in C57BL/6 mice via IV tail vein injection using IgG1 anti-GPIIb/IIIa (clone MWReg30), which mediates thrombocytopenia through FcγRIII (A), or IgG2a anti-GPIIb/IIIa (clone 5C4), which mediates thrombocytopenia through FcγRs but is not dependent on FcγRIII (B). Anti-CD44 (α-CD44) or isotype control was administered IV by tail vein injection 30 minutes before anti-platelet antibody injection. Platelet counts were determined at 24 hours after anti-platelet antibody administration. Data are from 6 (A) or 7 (B) independent experiments with 1 mouse per experiment. Error: mean ± standard error of the mean (SEM). ****P < .0001, 1-way analysis of variance with Tukey’s post hoc test.
The Fc region of anti-CD44 is required to ameliorate murine anti-GPIIb/IIIa antibody–mediated ITP
The Fc region of anti-CD44 was required to blockade macrophage FcγRs in vitro. Therefore, we evaluated the requirement of the anti-CD44 Fc region for the amelioration of murine ITP. IgG1 anti-CD44 was deglycosylated using PNGase-F or made into F(ab′)2 fragments. C57BL/6 mice were treated with equimolar quantities of the CD44 antibody variants before inducing anti-GPIIb/IIIa–mediated ITP. Anti-CD44’s ameliorative properties were completely lost when the Fc region was deglycosylated (Figure 7A) or removed [F(ab′)2] (Figure 7B), supporting the key role for the anti-CD44 Fc region observed in vitro for FcγR blockade. Because of the short half-life of F(ab′)2 antibodies, platelet counts were also evaluated at 1 and 2 hours post-GPIIb/IIIa antibody injection. Anti-CD44 F(ab′)2 did not have any effect at the 1- and 2-hour time points, whereas unmodified anti-CD44 mediated significant protection at all time points (supplemental Figure 7).
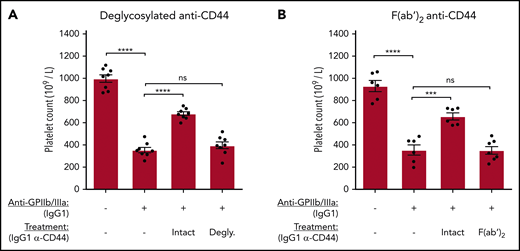
The Fc region of anti-CD44 is critically required to ameliorate murine ITP. Passive antibody-mediated ITP was induced in C57BL/6 mice using anti-GPIIb/IIIa IgG1 (clone MWReg30) administered IV by tail vein injection. IgG1 anti-CD44 or equimolar quantities of deglycosylated (Degly.) or F(ab′)2 variants were administered IV by tail vein injection 30 minutes before anti-platelet antibody injection. Platelet counts were determined at 24 hours after anti-platelet antibody administration using a Z2 Coulter Counter. (A) Mice treated with unmodified IgG1 (Intact) anti-CD44 (α-CD44) or its deglycosylated variant. (B) Mice injected with unmodified IgG1 (Intact) α-CD44 or its F(ab′)2 fragment. Data are from 4 independent experiments with 2 mice per experiment (8 mice total) (A) or from 6 independent experiments with 1 or 2 mice per experiment (6-7 mice total). Error: mean ± standard error of the mean (SEM). ****P < .0001, ***P < .001, 1-way analysis of variance with Tukey’s post hoc test.
The Fc region of anti-CD44 is critically required to ameliorate murine ITP. Passive antibody-mediated ITP was induced in C57BL/6 mice using anti-GPIIb/IIIa IgG1 (clone MWReg30) administered IV by tail vein injection. IgG1 anti-CD44 or equimolar quantities of deglycosylated (Degly.) or F(ab′)2 variants were administered IV by tail vein injection 30 minutes before anti-platelet antibody injection. Platelet counts were determined at 24 hours after anti-platelet antibody administration using a Z2 Coulter Counter. (A) Mice treated with unmodified IgG1 (Intact) anti-CD44 (α-CD44) or its deglycosylated variant. (B) Mice injected with unmodified IgG1 (Intact) α-CD44 or its F(ab′)2 fragment. Data are from 4 independent experiments with 2 mice per experiment (8 mice total) (A) or from 6 independent experiments with 1 or 2 mice per experiment (6-7 mice total). Error: mean ± standard error of the mean (SEM). ****P < .0001, ***P < .001, 1-way analysis of variance with Tukey’s post hoc test.
Discussion
The ability of anti-CD44 to mediate beneficial effects in autoimmune and inflammatory animal models has been well documented,1-10,25,31-36 including in numerous models in which IVIg is also effective.10 We previously proposed anti-CD44 as a potential IVIg alternative, particularly for ITP, in which anti-CD44 ameliorates disease similarly to IVIg in murine models but at a 3-log-fold lower dose.9,10 However, there is no mechanistic explanation for anti-CD44 activity in murine ITP, impeding development of anti-CD44 as a potential IVIg alternative. Here, we demonstrate that the anti-inflammatory activity of anti-CD44 in murine ITP is explained by inhibition of the FcγR pathway.
We observed that 2 anti-CD44 antibodies inhibited >90% of macrophage phagocytosis in 2 in vitro cytopenia models of IgG-opsonized platelets and IgG-opsonized RBCs. Phagocytosis inhibition required the anti-CD44 antibody Fc region, leading to blockade of the FcγR IgG binding site but without apparent changes in surface FcγR expression. Anti-CD44 did not lose significant effect when cell density was decreased to yield isolated macrophages, suggesting that blockade can occur via CD44 antibody coengaging with macrophage CD44 and FcγRs in cis. Further, anti-CD44 ameliorated murine passive ITP in a manner that is completely consistent with the mechanism of FcγR inhibition observed in vitro, with anti-CD44 acting in an IgG subclass–specific and Fc-dependent manner.
In the majority of inflammatory/autoimmune models in which anti-CD44 has efficacy, including arthritis, EAE, and experimental autoimmune uveoretinitis, its effect was explained by abrogating leukocyte extravasation to the site of inflammation.1,7,8,31 These effects are understood to be caused by the antigen-binding fragments of anti-CD44, and F(ab′)2 anti-CD44 can modulate leukocyte rolling similarly to intact anti-CD44.37 However, we now show that the Fc region of anti-CD44 mediates a separate anti-inflammatory mechanism that can prevent FcγR-mediated disease in vivo. Given that autoreactive antibodies also drive disease in anti-CD44–treatable murine models of arthritis,19,20 spinal cord homogenate-induced EAE,21 and passive ITP,9,10 part of anti-CD44’s beneficial effects in these models may be attributable to inhibiting FcγRs. Nedvetzki et al observed that the Fab′ fragment of anti-CD44 clone KM81 mediated reduced (∼50%) ameliorative activity compared with the intact antibody in the collagen-induced arthritis model,3 although this fragment also lacks CD44 crosslinking. CD44 signaling has been shown to involve spleen tyrosine kinase (Syk), a critical kinase in the signaling of FcγRs.38 Engagement of anti-CD44 to CD44 could lead to the recruitment of Syk, restricting its availability to FcγRs. However, to the extent that CD44 crosslinking and/or CD44 signaling by anti-CD44 contributed to FcγR inhibition, these effects were not sufficient alone, because F(ab′)2 anti-CD44 of either subclass failed to inhibit FcγR-mediated phagocytosis. Regardless, the total requirement of the anti-CD44 Fc region in ITP clearly demonstrates an anti-inflammatory mechanism mediated by this fragment of the antibody.
It is generally accepted that FcγRs play a major role in platelet clearance in human ITP.11 In particular, autoantibodies against GPIIb/IIIa are understood to drive FcγR-mediated clearance,11,13 although IgG glycoforms (which can alter FcR binding)39 appear normal in ITP.40 Therapies targeting FcγRs have shown efficacy in some ITP patients, including, most recently, a small molecule inhibitor of Syk (fostamatinib).17 Fostamatinib targets FcγRs by inhibiting Syk, which may undesirably inhibit a diverse array of other receptor and processes in which Syk is involved.41 However, in some cases, this “off-target” inhibition may be beneficial, because an apparent reduction in thromboembolic events has been observed in fostamatinib-treated ITP patients,42 potentially as a result of the role of Syk in platelet glycoprotein VI43 and platelet CLEC2 signaling.44 Currently, no blocking agent specific to FcγRs is approved for use. As presented here, anti-CD44 may be particularly suited for ITP as an IVIg alternative because of its significant FcγR-blocking effect and the major role that FcγRs play in ITP pathophysiology. Additionally, the apparent direct FcγR-blocking effect of anti-CD44 may block the effect of C-reactive protein, another FcγR ligand that has been shown to enhance platelet clearance in vitro and in vivo.45
Anti-CD44 has been shown to be capable of reversing disease in multiple murine autoimmune and inflammatory models,1,2,4,6,7 although we show a preventative murine ITP model in this study. Inhibition of FcγRs is a viable therapeutic strategy for human ITP,17,46,47 and anti-CD44 can also inhibit FcγR-mediated phagocytosis by human splenic macrophages (data not shown); however, future studies are needed to confirm the therapeutic potential in ITP. Development of a humanized anti-CD44 antibody (clone PF-03475952) for the treatment of autoimmunity (rheumatoid arthritis) was reported by Pfizer,48 but it was designed on the human IgG2 subclass.48 Our data presented here suggest that selection of the IgG2 subclass background may reduce anti-CD44 efficacy by removing potential FcγR inhibition, because IgG2 antibodies possess poor FcγR binding, including no binding to FcγRI,49 an important FcγR in autoimmune diseases, such as ITP.13 Human IgG1 may be a good candidate subclass to design anti-CD44 for FcγR inhibition, because IgG1 can bind all FcγRs.49 Although IgG3 can also bind all FcγRs, but with generally superior affinity to IgG1, which may provide superior FcγR inhibition, problems related to a shorter half-life and a large number of allotypes make IgG3 less desirable.50-52
The broad therapeutic concordance between IVIg and anti-CD44 led us to suggest anti-CD44 as a potential alternative to IVIg.10 Mechanistic similarities between the 2 interventions exist: anti-CD44 and IVIg do not require FcγRIIb10 or FcRn53 to ameliorate murine ITP. As presented here, anti-CD44 and IVIg share another potential mechanistic similarity with FcγR inhibition: a long-proposed explanation for IVIg activity.54 These mechanistic similarities may help to explain why IVIg and anti-CD44 share strong concordance as therapies for autoimmune and inflammatory models. Replacement of IVIg is of significant interest because it is a blood donor–derived product. Probably chief among the consequences of IVIg as a blood product is the significant issue of supply shortages. Monoclonal antibodies offer the potential to be produced at scale to demand. IVIg is also associated with undesirable side effects, such as thromboembolic events through several proposed mechanisms of increased blood viscosity,55 anti-cardiolipin antibodies,56 and the presence of contaminant procoagulant clotting factors present within some IVIg products,57 as well as hemolysis due to isohemagglutinin anti-A and anti-B antibodies.58
To our knowledge, the phenomenon of antibodies targeting surface antigens of FcγR-bearing cells mediating FcγR blockade was first described by Roger Kurlander.59 It was demonstrated that a variety of monoclonal antibodies against surface antigens of U-937 cells blocked human IgG1 binding in an Fc-dependent manner, an effect that did not lead to FcγR internalization and was stable in culture for ≥3 hours.59 We observed here that monoclonal antibodies against the highly expressed CD44 protein mediate significant FcγR inhibition through a similar mechanism: antibodies to CD44 appear to coengage with CD44 and FcγRs on the macrophage surface, mediating FcγR blockade without apparent loss of surface FcγR expression. Further, anti-CD44 functioned in complete concordance with this mechanism in vivo in the passive ITP model, although we acknowledge that we do not have definitive evidence that the in vitro and in vivo mechanisms are fully the same. Nonetheless, we demonstrate a novel anti-inflammatory mechanism of anti-CD44 that inhibits macrophage FcγR function and explains murine ITP amelioration. Our data support anti-CD44 as a potential IVIg alternative that may be of particular benefit in ITP because of the significant role that FcγRs play in human ITP pathophysiology.
All noncommercial materials will be made available to other investigators without any unreasonable restrictions. Detailed methodologies are provided in supplemental Materials and methods, and all protocols can be provided on request. Data sharing requests should be sent to Alan H. Lazarus (alan.lazarus@unityhealth.to).
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
This work was supported by the Canadian Blood Services Intramural Research Grant Program (A.H.L., H.N.), Canadian Institutes of Health Research Foundation grant 389035 (H.N.), and the Canadian Blood Services Graduate Fellowship Program (P.A.A.N., R.K.), funded by the federal government (Health Canada) and the provincial and territorial ministries of health.
The views herein do not necessarily reflect the views of the Canadian Blood Services or the federal, provincial, or territorial governments of Canada.
Authorship
Contribution: A.H.L. and P.A.A.N. conceived the study; P.A.A.N., G.K., and A.H.L. designed experiments; P.A.A.N., G.K., and R.K. performed experiments; P.A.A.N., G.K., R.K., and A.H.L. analyzed and interpreted data; G.Z. and H.N. provided vital reagents; P.A.A.N. and G.K. prepared the figures; P.A.A.N. wrote the manuscript; and P.A.A.N., G.K., R.K., H.N., and A.H.L. edited the manuscript.
Conflict-of-interest disclosure: A.H.L. holds patents on the use of monoclonal antibodies as replacements for IVIg and has received research funding from Rigel Pharmaceuticals and CSL Behring. G.Z. and H.N. have received research funding from CCOA Therapeutics. The remaining authors declare no competing financial interests.
Correspondence: Alan H. Lazarus, Keenan Research Centre, St. Michael’s Hospital, Unity Health Toronto, 209 Victoria St, Toronto, ON M5B 1T8, Canada; e-mail: alan.lazarus@unityhealth.to.