In this issue of Blood, 1 provide compelling evidence that resistance to Notch inhibitor therapy in early T-cell precursor acute lymphoblastic leukemia (ETP-ALL) occurs as a result of an activated phosphatidylinositol 3-kinase (PI3K) pathway. To further decipher the resistance mechanism, the investigators performed single-cell RNA sequencing analysis on the bone marrow of 5 patients treated with the γ-secretase inhibitor (GSI) BMS-906024 and found 13 different cell clusters, of which 6 were specific to leukemia patients.
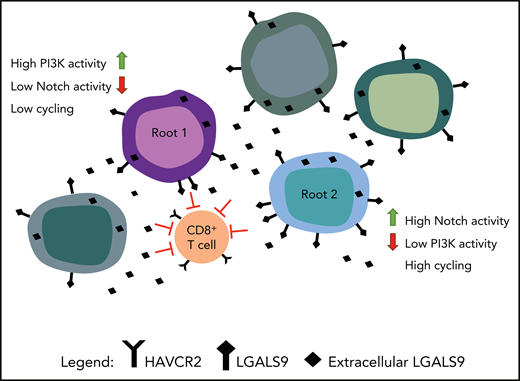
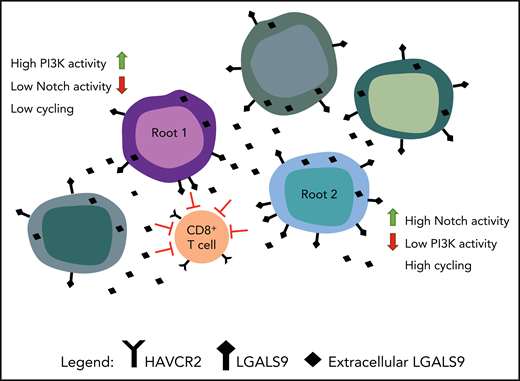
As shown in Amand et al, the demonstration of 2 different root cells generated from single-cell sequencing. Root cells are shown interacting with T cells through the expression of HAVCR2 (Galectin-9) and LGALS9 (TIM-3). The figure has been adapted from the visual abstract in the article by Anand et al that begins on page 2463.
As shown in Amand et al, the demonstration of 2 different root cells generated from single-cell sequencing. Root cells are shown interacting with T cells through the expression of HAVCR2 (Galectin-9) and LGALS9 (TIM-3). The figure has been adapted from the visual abstract in the article by Anand et al that begins on page 2463.
ETP-ALL is a high-risk subtype of T-cell acute lymphoblastic leukemia (T-ALL) characterized by high rates of relapse and induction failure with a unique immunophenotype, that is, the absence of CD4, CD8, and CD1a, and frequent expression of myeloid markers.2 Although this phenotype is the “gold standard,” Anand et al found that single leukemic cells express both strong stem cell signatures, and simultaneously, the most differentiated thymocyte markers. These findings suggest that the true phenotype of ETP-ALL is much more complex because of its intricate lineage plasticity. Earlier work suggested the absence of NOTCH1 mutations in ETP-ALL patients; however, recent whole-genome and transcriptome sequencing studies3,4 demonstrate that NOTCH1 mutations are present in a portion of ETP-ALL patients, providing the rationale for this trial of a GSI Notch inhibitor.5 However, clinical trials directly targeting NOTCH1 in T-ALL patients have not been highly successful, due in part to the rapid development of resistance to GSI.
Anand et al hypothesized that after GSI treatment, resistant stemlike cells might persist and then expand. Indeed, bioinformatics analysis identified 2 highly distinct stemlike “Root” states, low-cycling root 1 cells containing low Notch but highly activated PI3K pathway, and fast-cycling root 2 cells containing high Notch levels but low PI3K activation (see figure). After GSI treatment, the researchers found that PI3K levels were inversely correlated with Notch levels. Interestingly, 1 of the 5 patients analyzed had a complete response (CR) to GSI,5 even though the patient’s marrow contained root 1 cells. Did this CR occur because there were fewer root 1 cells in the bone marrow or did other genes play a role? For example, the Pim kinase, which collaborates with PI3K/AKT, can be induced when Notch activity is inhibited.6 The MAPK, TNF-α, and NF-κB signaling pathways are shown to be positively correlated with Notch levels in root cells. GSI treatment, although suppressing Notch activity, leaves these signals intact, thus available to collaborate with the PI3K pathway and further enhance GSI resistance.
Anand and colleagues also demonstrate that a broadly active PI3K inhibitor Buparlisib when combined with a GSI effectively inhibits T-ALL cell growth in vitro and blocks GSI-resistant clones. This result is consistent with the fact that root 1 cells with high PI3K and low Notch exist prior to GSI therapy. However, given the number of pathways available that permit resistance, a more comprehensive approach is likely needed. Previously, T-ALL cells resistant to GSI (persisters) were shown to be sensitive to the BRD4 inhibitor JQ1.7 This agent has a broad spectrum of action, including inhibiting Myc transcription and mTOR signaling,7 as well as inhibiting other genes containing a superenhancer, including Pim. JQ1 has also been shown to decrease activated AKT levels.8 Therefore, the addition of this class of agents alone or combined with a PI3K/AKT inhibitor may prevent resistance to Notch inhibition.
The investigators suggest that another resistant population of more mature leukemia cells exists that may escape Notch/PI3K inhibition based on transcribed genes that drive the immune system. CD8+ T cells in leukemic patients have markers of T-cell exhaustion with TIM-3 (HAVCR2) expression being prominent. All leukemic cells expressed Galectin-9 (LGALS9), the TIM-3 ligand, suggesting that leukemic cells could be activating this T-cell protein (see figure). In vitro CD8+ T cells exposed to T-ALL culture supernatant showed upregulation of the exhaustion markers TIM-3 and TIGIT. Interestingly, TIM-3 appears to be upregulated in a PI3K/AKT–dependent manner,9,10 for example, in tumor infiltrating lymphocytes in head and neck cancer. This suggests that targeting the PI3K/AKT pathway as part of the resistance mechanism will also in part block the TIM-3 pathway.
The Children’s Oncology Group protocol AALL1231 for the treatment of T-ALL includes long-term, very intensive, high-dose chemotherapy with and without bortezomib. Relapsed recurrent T-ALL is highly chemotherapy resistant and has an extremely poor prognosis. As shown by Anand et al, single-cell RNA sequencing could further explain the nature of resistance in these patients. By defining and then attacking the roots of resistance in Notch mutant cases, it may be possible to combine a GSI along with PI3K/AKT, or BRD4 inhibitor, with or without TIM-3 inhibitory antibody to rescue these patients. Future trials will certainly explore the treatment based on the avenues of resistance identified in the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.