Abstract
The retinoic acid receptors (RARA, RARB, and RARG) are ligand-regulated nuclear receptors that act as transcriptional switches. These master genes drew significant interest in the 1990s because of their key roles in embryogenesis and involvement in a rare malignancy, acute promyelocytic leukemia (APL), in which the RARA (and very rarely, RARG or RARB) genes are rearranged, underscoring the central role of deregulated retinoid signaling in leukemogenesis. Several recent provocative observations have revived interest in the roles of retinoids in non-APL acute myeloid leukemia (AML), as well as in normal hematopoietic differentiation. We review the role of retinoids in hematopoiesis, as well as in the treatment of non-APL AMLs. From this perspective, broader uses of retinoids in the management of hematopoietic tumors are discussed.
Introduction
Nuclear receptors are transcription factors with activities that can be regulated by a variety of small molecules. These master genes raise fascinating biological questions, notably with respect to their roles in developmental programs or adaptive responses. They also open major therapeutic avenues in multiple pathologies in which they are directly or indirectly implicated. One subgroup, retinoic acid receptors (RARs), is particularly relevant to the field of hematopoiesis, as 2 of the 3 receptors, RARA and RARG, have been implicated as key regulators of normal and transformed blood cells. Moreover, acute promyelocytic leukemia (APL) is driven by fusion genes involving RARs, primarily RARA.1 Remarkably, APL is clinically exquisitely sensitive to all-trans-retinoic acid (ATRA), the natural ligand of the RARs, exemplifying one of the best examples of targeted therapy, and (at least when combined with arsenic trioxide), the only rapidly and definitively curative one. The physiopathology of APL and its basis for response to therapy has been reviewed elsewhere.2,3 The APL model highlights 2 important ideas discussed here in the context of both normal hematopoiesis and non-APL acute myeloid leukemias (AMLs): (1) RARs and ATRA signaling are important for stem cell self-renewal and hematopoietic differentiation; and (2) ATRA therapy can have profound impacts on the outcomes of hematopoietic malignancies.
Deciphering ATRA signaling
The cloning of the RARs gave a molecular basis to ATRA signaling and prompted substantial efforts to understand its complex developmental and physiological impacts.4 In brief, ATRA signaling relies on 3 receptors: RARA, RARB, and RARG, as well as their retinoid X receptor (RXR) heterodimeric partners. Each receptor is expressed as several spliced isoform variants. ATRA is synthetized locally, through very complex mechanisms involving capture of retinol from the circulation, oxidation into retinal and retinoic acid, transport and sequestration, and, finally, degradation.5-7 Most, if not all, of the enzymes involved in retinoid metabolism are transcriptionally regulated by ATRA, ensuring, through local feed-forward and feed-back controls, the fine spatiotemporal tuning of ATRA abundance. Initially, simple models in which ligand-bound RARs complexed to DNA response elements to promote transcriptional activation of target genes prevailed.8 Yet, over the years, our knowledge of the molecular mechanisms involved has become more and more complex (Figure 1). RARA seems to exert primarily ATRA-reversible basal repressive functions, whereas RARG is generally a potent ligand-dependent transcriptional activator.9 Thus, different RARs may have antagonistic effects on shared targets. ATRA exposure can yield target transcriptional activation and initiate proteasome-dependent receptor degradation.10-12 Ligand-activated repression is well established for some targets, such as FGF8, which play a critical role in developmental pathways.5,7 Receptor activity can be regulated by posttranslational modifications, with functional consequences for embryonic stem cell (ESC) differentiation.13 Physiologically, basal repressive functions play a key role in development (especially in head formation in xenopus and mouse skeletal development),14 so that receptor loss or extinction can be quite different from vitamin A deprivation. Use of synthetic ligands can promote a variety of interactions between specific RARs and coactivator or corepressor proteins, broadening the diversity of molecular and cellular responses. Retinoic acid signaling can also be modulated by RXRs and their poorly characterized natural ligands, which seem to be important in hematopoietic cells.15 However, RXR signaling controls a much broader network, including the vitamin D3 receptor and other type 2 nuclear receptors, which also play critical roles in the pathophysiology of hematopoiesis and AML.16 This multiplicity of cross-connected effects stresses the importance of “clean” gene ablation experiments (or even ablation of response elements within target genes) in a physiological in vivo setting, rather than use of agonist or antagonist receptor ligands or dominant negative receptor expression in cell lines, to explore physiological questions.7
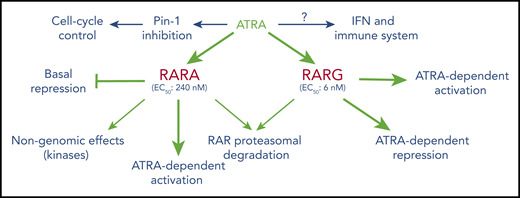
Overall effects of ATRA on RAR-mediated signaling. The data stress the diversity of mechanisms (basal repression, ligand-induced repression or activation, RAR degradation, cross talk with other key pathways) as well as important differences in ATRA levels implicated in these effects. In the hematopoietic lineage, RARA seems primarily involved in granulocytic differentiation, whereas RARG plays important roles in the biology of HSC and theirs niches.
Overall effects of ATRA on RAR-mediated signaling. The data stress the diversity of mechanisms (basal repression, ligand-induced repression or activation, RAR degradation, cross talk with other key pathways) as well as important differences in ATRA levels implicated in these effects. In the hematopoietic lineage, RARA seems primarily involved in granulocytic differentiation, whereas RARG plays important roles in the biology of HSC and theirs niches.
ATRA and RARA have also been implicated in imperfectly understood “nongenomic effects,” wherein RARA and ATRA effects are mediated outside of the nucleus, through modulation of protein kinase activity.17 Conversely, several drugs that target kinases modulate the effects of ATRA in AML cells (Table 1).18-20 Synergistic target gene activation and subsequent differentiation were reported with myelomonocytic growth factors (granulocyte and granulocyte-macrophage stimulating factor [G/GM-CSF]) and were thought to rely on activation of the mitogen-activated protein kinase (MAPK) pathway.21 Interestingly, upon administration of ATRA, MAPK can rapidly phosphorylate RARs and several associated proteins (corepressors, coactivators) such that this synergy may ultimately converge on ATRA target genes.20,22 In addition, activation of other kinases (including cyclin-dependent ones) targets not only RARA, but also coactivators and repressors.23 Finally, the notion that all ATRA-triggered effects necessarily occur through interactions with RARs has been challenged. Indeed, ATRA was shown to potently inhibit the peptidyl-prolyl cis-trans isomerase PIN-1 enzyme, which posttranslationally modifies a large number of proteins to promote growth.24-26 Importantly, PIN-1 is often activated in AMLs, such that its targeting by ATRA can impair growth.27 Collectively, these examples point to the versatility and complexity of the interplay between RARs, RXRs, and their ligands for the control of multiple biological processes. Although direct linking of a specific molecular mechanism to a given cellular phenotype has often been challenging, terminal granulocytic differentiation is thought to be RARA inhibited and ATRA/RARA activated,28 neuronal differentiation of ESCs requires a specific RARG isoform,13 and nongenomic RARA functions have recently been implicated in T-cell activation.29
ATRA signaling in normal hematopoiesis
An elegant study has demonstrated that RAR signaling is essential for the emergence of mouse hematopoietic stem cells (HSCs) from the hemogenic endothelium, through activation of WNT signaling.30 After completion of embryonic development, vitamin A, the precursor to ATRA, promotes HSC dormancy in adult mice,31 which is a likely explanation for the ability of ATRA to enhance the long-term repopulating activity of HSCs32 and protect them from exhaustion after stress-induced proliferative activation by interferons (IFNs). Conversely, deprivation of vitamin A induces a progressive loss of HSCs, impairs efficient restoration of HSC dormancy after inflammatory stress and promotes myeloid expansion.33 These ATRA/HSC connections illustrate one of the broad roles of retinoid signaling on multiple types of stem cells in vivo.7 Some master regulators of hematopoietic differentiation are under direct or indirect transcriptional control of retinoids, including KIT, CEBPA, WNT, and HOX genes.31 The HSC niche or, more broadly, the HSC microenvironment, appears to protect HSCs from pharmacological doses of ATRA,34 at least in part, through the expression of ATRA-degrading cytochromes.35 Downstream of HSCs, retinoids favor the emergence, growth, and differentiation of normal early committed myeloid progenitors with granulocytic potential, at the expense of erythroid and mastocytic ones36,37 (Figure 2). Overall, RA signaling is complex, depending on the stage of differentiation and drug concentrations, with low physiological levels and pharmacological doses occasionally conveying different signals.
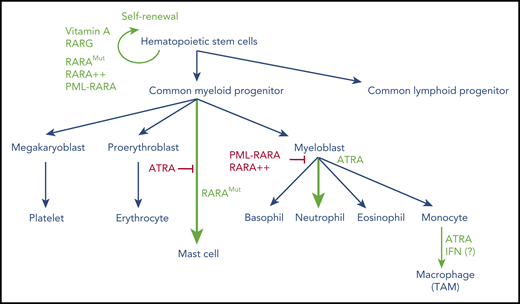
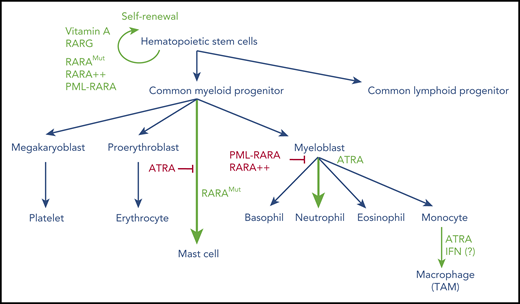
Overview of hematopoietic cell differentiation and impact of retinoid signaling. Green denotes activation; red denotes inhibition. RARA++, overexpressed RARA; RARAMut, mutated RARA; TAM, tumor-associated macrophage.
Overview of hematopoietic cell differentiation and impact of retinoid signaling. Green denotes activation; red denotes inhibition. RARA++, overexpressed RARA; RARAMut, mutated RARA; TAM, tumor-associated macrophage.
Molecularly, the contributions of individual RARs to hematopoiesis were first explored in RAR-deficient mice, demonstrating that basal granulocytic differentiation was accelerated not only by RARA excision, but also by pharmacological doses of ATRA solely in the presence of RARA.28 Thus, under physiological conditions, endogenous RARA slows down granulopoiesis. RARA is expressed from 2 alternative promoters, yielding isoforms with different N termini. These are differentially expressed during hematopoietic differentiation, with RARA-2 being massively induced upon myeloid differentiation.38 Therapeutically, acceleration of terminal differentiation by pharmacological concentrations of retinoids or RARA-specific agonists has been proposed in settings where functional granulocytes are limiting.39,40
RARA is a low-efficacy receptor for ATRA (50% effective concentration [EC50] 240 nM), whereas RARG very avidly binds its ligand and activates target genes (EC50 6 nM) and accordingly plays a key role in embryonic development.41,42 Importantly, RARG appears to be a physiological regulator of HSC self-renewal.43 Remarkably, this effect also involves the microenvironment, as RARG ablation within this niche suffices to induce a myeloproliferative syndrome, even when RARG expression is retained in reintroduced wild-type HSCs.44 More studies are needed to decipher the respective roles of HSCs and the niche in RARG effects. RARG and retinoids facilitate conversion of fibroblasts to induced pluripotent stem cells, again underscoring their tight relationships with stem cell differentiation.45 RARG expression and a specific polymorphism have also been linked to doxorubicin-induced cardiotoxicity.46,47
In contrast to RARA loss, dominant negative versions of RARA dramatically alter hematopoietic cell fate in several ways (reviewed by Collins36 ). First, these mutants may immortalize very early progenitors that are subsequently capable of multilineage differentiation (eg, toward B cells). Second, when expressed in a multipotent cell line, dominant negative RARA shifts spontaneous differentiation toward mastocytes, while abolishing the basal myelomonocytic fate. Moreover, when these progenitors are forced toward granulocytic lineage differentiation by exposure to GM-CSF, they undergo a promyelocytic differentiation block, which can be reversed by ATRA. In these conditions of overexpression, the dominant negative RARA mutant may perturb normal signaling of multiple other nuclear receptors (eg, by titrating their common RXR partners). Mysteriously, mere overexpression of normal RARA suffices to induce myeloid progenitor immortalization, as opposed to RARB or RARG.48 Altogether, through a variety of complementary mechanisms, retinoids and their cognate receptors exert multiple effects at various stages of hematopoietic differentiation: on self-renewal of very early progenitors (primarily RARG), mastocyte lineage commitment, and myeloid differentiation (primarily RARA; Figure 2).
ATRA-induced differentiation in AML
Historically, one of the first models of ATRA-induced differentiation was the AML cell line HL60, which differentiates into apparently normal granulocytes. Why these cells lacking the PML-RARA fusion are so exquisitely ATRA sensitive remains unresolved. Whereas ATRA-resistant HL60 cells harbor RARA mutations, demonstrating that RARA is the actual effector of ATRA activity in that setting, the key downstream targets remain largely unknown. Multiple studies have since demonstrated that, similar to normal progenitors, growth and differentiation of some primary AML blasts are, at least in part, ATRA- or RARA-agonist sensitive.49,50 Similar findings were made with ligands specific for RXRs alone, or in the presence of cAMP which allows transcriptional activation from RAR/RXR heterodimers.51,52
Most, if not all, of these initial studies used morphological differentiation as an end point. For decades, the field considered that terminal differentiation necessarily entailed loss of clonogenic activity. Yet, recent studies in APL or AML models have demonstrated that cancer cells profoundly engaged in the differentiation pathway can nevertheless revert to immature cells and regain clonogenic features.53-56 These provocative observations cast some caution on conclusions drawn from many previous findings. Thus, rather than terminal granulocytic differentiation, ATRA-induced loss of clonogenic activity or clear-cut prolongation of survival in vivo are the end points that are more likely to adequately reflect durable therapeutic efficacy in patients with AML. Although loss of leukemia-initiating cell activity may be envisioned as a specific form of differentiation, it is mechanistically distinct and may be uncoupled from terminal granulocytic maturation.55 While there are examples where retinoids modestly prolong survival of AML models or patient-derived xenografts,50 AML remission, not to mention eradication, is never achieved by ATRA alone, in sharp contrast to APL.
The natural vitamin A derivative used in most studies is ATRA, which activates all 3 RARs isoforms (although with different efficiencies; Figure 1). Receptor selectivity of drugs provides an interesting angle to approach their respective role. Intriguingly, a study found opposing effects of ATRA and a RARA-specific agonist for clonogenic growth of AML/ETO-transformed progenitors, suggestive of antagonist functions of RARs α and γ isoforms.57 The results of these experiments suggest that, in this model, ligand activation of RARA signaling efficiently promotes myeloid differentiation and loss of self-renewal, whereas ATRA, through RARG activation, may enhance self-renewal44 and/or decrease retinoid levels by activating cytochromes implicated in their catabolism.34,35 However, the respective roles of RARA and RARG in AML terminal differentiation and clonogenic activity remain open areas of investigation. Recent studies have explored tamibarotene, a RARA/RARB-specific agonist with a better pharmacodynamic profile than ATRA (in particular, a longer half-life).58 Tamibarotene, also known as SY-1425, can induce differentiation and initiate clinical responses in some clinically ATRA-resistant patients with APL, demonstrating its biological activity.59 When used in maintenance therapy after ATRA/chemotherapy APL treatment, it is slightly more efficient than ATRA in preventing late relapses,60 particularly in patients with FLT3 mutations, which antagonize ATRA signaling.19,61 Tamibarotene can also efficiently promote differentiation of some AML cells ex vivo and in vivo, and this effect has been linked to existence of an RARA superenhancer and monocytic differentiation.50
Studies in ESCs have revealed that differentiation responses are intimately regulated by the cell cycle,62 possibly explaining the synergistic effects of ATRA with many drugs favoring S-phase arrest of AML cells. The modest in vivo antileukemia effects of ATRA alone (despite its ability to initiate differentiation in AML) stresses the importance of combinations with other drugs. In that respect, a broad repertoire of drugs enhances the effects of ATRA to block AML proliferation or promote differentiation (Table 1), with sometimes additive survival effects in xenograft models or even in patients. Whether this represents additive effects of parallel pathways or rather convergence of distinct pathways onto RAR signaling is currently not known. Through induction of cell cycle arrest, ATRA-induced Pin-1 inhibition may synergize with “classic” RAR activation when using pharmacological treatments with very high doses of ATRA, as in AML therapy. Importantly, synergies between retinoids and DNA hypomethylating agents have been reported recently in patients with AML.63
ATRA signaling in genetically defined AML models
Recent studies have tried to genetically define ATRA-sensitive AMLs50,64 and explore the reciprocal roles of AML oncogenes and ATRA signaling, but an unambiguous picture has not emerged as yet. Many of these correlative studies were performed in cell lines rather than in primary patient cells and were used differentiation as an end point. The first set of studies focused on NPM-1c–associated AMLs, because these initially appeared clinically to be ATRA sensitive.65,66 Similar to its role in APL, ATRA may initiate blast differentiation in these AML cells, along with activation of the TP-53 pathway and degradation of mutant NPM-1c,67,68 a protein required for maintenance of the transformed state.69 However, NPM-1c degradation may not fully recapitulate ATRA effects in theses AML cell lines (A. Bazarbachi, Beirut, Lebanon, written communication, 23 November 2020).
Other important AML oncogenes are the isocitrate dehydrogenase (IDH) enzymes, and some studies have shown correlation of ex vivo ATRA efficacy with their mutation.26,70 Several studies have suggested that reactive oxygen species (ROS) basally induced by IDH mutation facilitate ATRA-induced AML differentiation by inhibiting lysine demethylase 1A (LSD-1/KDM1A) activity. The LSD-1 histone demethylase impedes ATRA response in AML,71 as well as in some normal cells.72 Accordingly, LSD-1 inhibition favors ATRA-induced AML differentiation,71,73 a concept that is currently being investigated in patients.74 Intriguingly, mechanistic studies have demonstrated that LSD-1 inhibitors act to sensitize AML cells to ATRA-induced differentiation by impeding LSD-1 interaction with growth factor independent 1 transcriptional repressor (GFI-1),75 rather than through inactivation of its histone demethylase enzymatic activity. Thus, in AML, as in medulloblastoma,76 LSD-1 and GFI-1 cooperate to enforce transformation, but, at least in AML, LSD-1 has a scaffolding, rather than an enzymatic, function. GFI-1 also appears to be a critical factor in controlling ATRA response and granulocytic differentiation in APL.77 Overall, GFI-1 may be a master prodifferentiation transcription factor downstream of ATRA-induced, LSD-1–repressed signaling, in several different tumors, including AML.78 Pharmacological induction of ROS through iron deprivation was also proposed to promote differentiation through enhanced VitD-3 signaling79,80 or control of global sumoylation, which facilitates ATRA-induced differentiation.81
Significant evidence, in APL and other AMLs, has suggested that FLT3 activation precludes efficient ATRA signaling and that clinically used FLT3 inhibitors can rescue efficient ATRA-induced differentiation and clinical responses.19,61,82 Other studies have linked expression of EVI1 with ATRA sensitivity.83-86 At the target gene level, EVI-1 can indeed enhance or inhibit activation of various ATRA target genes. However, EVI-1 sensitizes to ATRA-induced differentiation, whereas ATRA enhances the leukemia stem cell (LSC)-promoting activities of EVI-1, so that the overall clinical benefit of ATRA in these malignancies may be complex.64 MN1 is an oncogene in AML that regulates RARA signaling. It has been suggested that high MN1 expression is associated with ATRA responsiveness,87,88 but this remains to be further explored. Apart from direct oncogenic drivers, molecules with deregulated expression in malignancies such as PRAME89,90 were proposed to blunt ATRA response. How this relates to AML biology and resistance to ATRA signaling remains to be established.
ATRA therapy in patients with AML
Some trials initially suggested that ATRA could benefit AML therapy when added to standard anthracycline/cytarabine–based AML regimens.65,66 Patients with good prognoses who bear the NPM-1c mutation, but not FLT3 pathway activation, seem to benefit the most from ATRA adjunction.65,91 Unfortunately, these results were not confirmed by other groups, and so confusion prevails.92-94 Other clinical studies explored the addition of ATRA and/or valproic acid (a histone deacetylase inhibitor) to chemotherapy and observed a decrease in late relapses in elderly patients receiving ATRA and chemotherapy95,96 (discussed later). In another study, the combination of ATRA, VitD-3 and low-dose maintenance chemotherapy yielded a significant survival advantage in AML.97
Most often, AML is a disease which occurs after 70 years of age, when many patients are unfit for cardiotoxic anthracycline-based therapies. This has raised considerable interest in nongenotoxic approaches based on “biomodulators,” often a combination of differentiation inducers with histone deacetylase inhibitors and DNA demethylating agents, all of which are predicted to facilitate re-expression of differentiation genes repressed by AML oncogenes. Combinations of ATRA and valproic acid without chemotherapy initiated ex vivo differentiation, but yielded disappointing results in patients.98,99 In contrast, when ATRA was combined with the hypomethylating agent decitabine, unambiguous enhancement of decitabine responses were observed in elderly patients with AML.63 Essentially similar results were very recently reported with tamibarotene and azacytidine in unfit patients with AML, responses being more frequent in RARA-overexpressing AMLs (registered on www.clinicaltrials.gov as #NCT02807558),100 pressing for a direct comparison between the potencies of ATRA and tamibarotene in this setting. Simultaneous or sequential drug administration schemes may also bear some importance in long-term efficacy. Mechanistically, a study suggested that hypomethylating agents and RARA-specific retinoids can synergize to induce apoptosis of AML cell lines.101 Clinically, in the absence of clear physiopathological mechanisms and simple biomarkers, identification of intrinsically ATRA-sensitive AMLs has remained an unmet challenge. Yet, the clinical results of the decitabine/retinoid combinations raise a question as to whether there is any beneficial role for the addition of retinoids (ATRA or tamibarotene) to the current decitabine/venetoclax standard for unfit patients with AML.102
A striking observation was recently reported in 2 children morphologically and immunophenotypically diagnosed with APL and accordingly treated with a frontline ATRA-chemotherapy APL regimen. Remarkably, in these patients, the RARA gene was mutated at relapse, although extensive molecular studies failed to demonstrate any RAR fusions.103 These somatic RARA mutations acquired during ATRA therapy are known to confer dominant negative effects, similar to those found in rare forms of breast cancers.104 This unheralded, but key, clinical observation implies that heavy exposure to ATRA (both induction and maintenance regimen) and chemotherapy exerted a potent, RARA-dependent, negative selective pressure on the leukemic clone (Figure 3). These definitive observations could further renew interest for ATRA-containing regimens in defined AML subsets, as discussed earlier. Overall, this field of investigation remains active, and exciting discoveries may be expected, particularly if combination therapy for AML with ATRA and DNA demethylating agents yields unambiguous survival advantages and reliable biomarkers for ATRA sensitivity are identified.
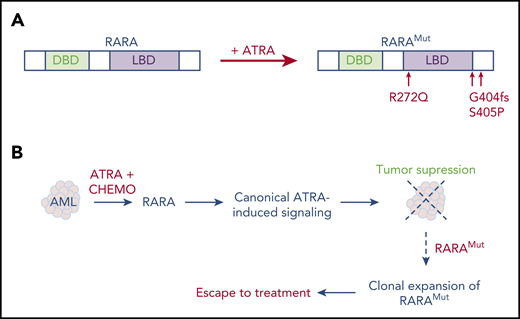
Sequential dual effects of ATRA therapy in patients with AML. (A) Selection of RARA mutations conferring ATRA resistance in ATRA-treated pediatric AML,102 fs, frameshift. (B) Proposed mechanism for ATRA activity and mutant selection in ATRA-treated AML. Treatment-selected dominant-negative RARA mutants (RARAMut) enhance self-renewal and block differentiation, contributing to AML relapses. CHEMO, anthracycline-based chemotherapy; LBD, ligand-binding domain; DBD, DNA-binding domain.
Sequential dual effects of ATRA therapy in patients with AML. (A) Selection of RARA mutations conferring ATRA resistance in ATRA-treated pediatric AML,102 fs, frameshift. (B) Proposed mechanism for ATRA activity and mutant selection in ATRA-treated AML. Treatment-selected dominant-negative RARA mutants (RARAMut) enhance self-renewal and block differentiation, contributing to AML relapses. CHEMO, anthracycline-based chemotherapy; LBD, ligand-binding domain; DBD, DNA-binding domain.
Future prospects for retinoids in treatment of hematopoietic malignancies
APL was considered for decades to be the premier model for differentiation therapy.54,105 Yet, it is now clear that differentiation is not the main contributor to the clinical efficacy of ATRA or arsenic and that terminal AML differentiation can spontaneously reverse.2,56 When exploring AML sensitivity to retinoids, the clinical expectations were drawn from the APL models and were very likely much too high.54 The dramatic ATRA activity in APL reflects PML/RARA degradation and requires reformation of PML nuclear bodies initially disrupted by expression of the fusion protein.106,107 Nuclear body reformation activates a PML/P53 senescence checkpoint that enforces loss of self-renewal,2 which explains the exquisite therapy sensitivity of PML/RARA–driven APLs2 and, because arsenic targets PML, the ATRA/arsenic resistance of X-RAR–driven variant APLs.1,53,108 Interestingly, in neuroblastoma as in AML, efficiency of retinoids in mouse models was boosted by decitabine.63,101,109 Decitabine may activate senescence or loss of self-renewal by activating expression of transposable elements and IFN secretion,110 a process enhanced by retinoids, and possibly implicating PML as a downstream effector.111 Thus, AML response to the retinoid/decitabine combination may somehow mechanistically resemble APL response, implicating PML, as does IFN/arsenic activity in myeloproliferative neoplasms.112 Paradoxically, it is possible that the concept of differentiation therapy actually better applies to AML than to APL. These may resemble neuroblastoma, where 13-cis-retinoic acid has significant (although modest) clinical utility as maintenance therapy, presumably by promoting differentiation and loss of self-renewal of residual neuroblastoma-derived neuronal precursors.113 This concept of retinoid-based maintenance may actually apply beyond AML and neuroblastoma. The broad effects of ATRA on growth control and LSC status may have clinical usefulness in other hematological malignancies, such as acute lymphocytic leukemia.114
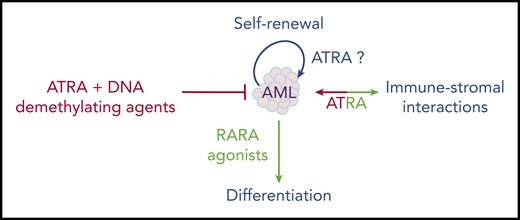
One recently reexplored axis is the interplay between ATRA and the immune system, notably IFN signaling.115 A RARA/STAT1 pathway was proposed to explain how ATRA enhances IFN target genes.116 Moreover, the ATRA-regulated RIG-1 gene controls basal IFN signaling.117 This RARA/IFN cross talk was explored in a liver cancer model resulting from loss of Tif1a, wherein mere Rara haploinsufficiency was sufficient to abrogate tumor development.118 In that context, Tif1a loss was associated with massive induction of IFN signaling in developing hepatocytes, which, similar to transformation in vivo, was reversed by loss of a single Rara allele.119 This model underscores the key, but mechanistically still poorly understood, connections between RARA and IFN-pathway activation. Accordingly, in the immune system, retinoids modulate dendritic cell differentiation120 and bacterial quorum sensing.121 Some of the ATRA effects in cancers may reflect not only cell-autonomous pathways, but also the interplay between the niche and the immune stroma (Figure 4). In that respect, tumor-derived ATRA has been proposed to orient the microenvironment toward an immune-suppressive phenotype, and RARA antagonists proposed to facilitate the activity of checkpoint inhibitors.122 Similarly, in chronic lymphocytic leukemia (CLL), leukemic cells induce ATRA synthesis in the stromal microenvironment to favor leukemia expansion.123 This emerging role of ATRA in the microenvironment may be important in the context of ATRA/chemotherapy combinations in AMLs.
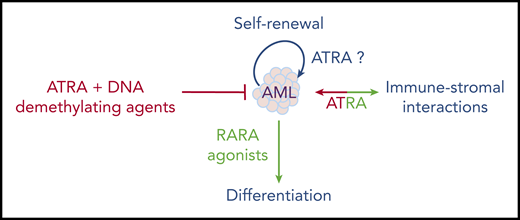
Overall, although many aspects of retinoid physiopathology remain incompletely understood, they have unexpectedly emerged in diverse aspects of medicine. As in APL, clinical breakthroughs often occur by chance and the mechanics are only later understood. The multiple facets of ATRA activity in normal hematopoiesis do not yet allow for a simple prediction of its therapeutic effects in leukemia; moreover, the emerging roles of ATRA in normal and stromal niche interactions further complicate these issues. Therapeutically, these are exciting times: after the APL explosion, followed by a window of boredom and neglect, retinoids may be revived into efficient “à la carte” combinatorial therapies based on sound physiopathological explorations.
Acknowledgments
The authors thank V. Lallemand-Breitenbach, J. Soulier, H. Dombret, E. Laplantine, A. Bazarbachi, Christopher Bassil, and laboratory members for advice and comments on the manuscript, and F. Maloumian for the artwork.
Work in the authors' laboratory is supported by INSERM, Centre National de la Recherche Scientifique (CNRS), Paris, Sciences, Lettres (PSL) University, the CAMELIA Project from Institut National du Cancer (INCA), Foundation Saint Michel, European Research Council Advanced Grant 785917–PML-THERAPY and the European Research Area Network on Translational Cancer Research (TRANSCAN) DRAMA Project.
Authorship
Contribution: All authors wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Hugues de Thé, Collège de France, Place Marcelin Berthelot, 75005 Paris, France; e-mail: hugues.dethe@inserm.fr.