Key Points
NSC12 induces antitumor activity in WM.
NSC12-dependent FGF blockade targets WM cells via MYD88 inhibition.

Abstract
The human fibroblast growth factor/fibroblast growth factor receptor (FGF/FGFR) axis deregulation is largely involved in supporting the pathogenesis of hematologic malignancies, including Waldenström macroglobulinemia (WM). WM is still an incurable disease, and patients succumb because of disease progression. Therefore, novel therapeutics designed to specifically target deregulated signaling pathways in WM are required. We aimed to investigate the role of FGF/FGFR system blockade in WM by using a pan-FGF trap molecule (NSC12). Wide-transcriptome profiling confirmed inhibition of FGFR signaling in NSC12-treated WM cells; unveiling a significant inhibition of MYD88 was also confirmed at the protein level. Importantly, the NSC12-dependent silencing of MYD88 was functionally active, as it led to inhibition of MYD88-driven pathways, such as BTK and SYK, as well as the MYD88-downstream target HCK. Of note, both canonical and noncanonical NF-κB cascades were downregulated in WM cells upon NSC12 treatment. Functional sequelae exerted by NSC12 in WM cells were studied, demonstrating significant inhibition of WM cell growth, induction of WM cell apoptosis, halting MAPK, JAK/STAT3, and PI3K-Akt pathways. Importantly, NSC12 exerted an anti-WM effect even in the presence of bone marrow microenvironment, both in vitro and in vivo. Our studies provide the evidence for using NSC12 as a specific FGF/FGFR system inhibitor, thus representing a novel therapeutic strategy in WM.
Introduction
The human fibroblast growth factor/fibroblast growth factor receptor (FGF/FGFR) axis plays an essential role in a wide range of cellular processes, such as cell proliferation, differentiation, migration, and survival, and its oncogenic role has been described in both solid tumors and hematologic malignancies, including multiple myeloma and Waldenström macroglobulinemia (WM).1-10 WM is a low-grade lymphoproliferative disorder characterized by the presence of a lymphoplasmacytic infiltrate in the bone marrow (BM), serum monoclonal immunoglobulin M,11,12 and a somatic mutational profile involving MYD88 and CXCR4 that has been shown to support WM pathogenesis and disease progression.13-19 Nevertheless, WM is still an incurable disease, and patients succumb because of disease progression.11-15,17-23 Therefore, novel therapeutics designed to specifically target deregulated signaling pathways in WM are required. We aimed to investigate the role of the FGF/FGFR system in WM by using a pan-FGF trap molecule (NSC12), responsible for FGF/FGFR blocking, given the overexpression of FGFR3 in tumor cells, as compared with their normal cellular counterpart.4 There have been studies reporting on the use of the multireceptor tyrosine kinase (RTK) inhibitor, benzimidazolyl-quinolinone dovitinib, as an anti-WM approach.4 Dovitinib, a benzimidazole-quinolinone, inhibits the phosphorylation of several type III to V RTKs, such as vascular endothelial growth factor receptor, platelet-derived growth factor receptor, FMS-like tyrosine kinase 3 (FLT3), stem cell factor receptor (c-KIT), colony stimulating factor receptor 1, and FGFRs.10,24-26 Therefore, there is a need to develop novel therapeutic interventions designed to specifically neutralize the FGF/FGFR axis, aiming to avoid toxicities because of the off-target effects secondary to RTK inhibition. NSC12 has been recently identified as a pentraxin-3–derived small molecule and developed as an oral pan-FGF trap,27,28 able to silence the FGF/FGFR signaling cascade, leading to an antineoplastic activity in those tumors where FGF/FGFR activation plays an oncogenic role.
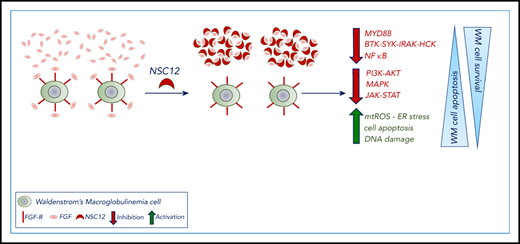
Here, we present the antineoplastic activity exerted by NSC12, as a result of the specific FGF/FGFR targeting within the context of WM where a therapeutic strategy designed to specifically halt the FGF/FGFR axis has not been reported so far. By investigating the underlying molecular mechanisms of NSC12-dependent anti-WM effect, we unveil how the NSC12-dependent FGF/FGFR silencing leads to MYD88 inhibition coupled with blocking of the canonical and noncanonical NF-κB pathway, and other prosurvival cascades, including PI3K-AKT, MAPK, and STAT3. Overall, the NSC12-dependent modulation of these signaling cascades ultimately resulted in inhibition of WM tumor growth, arrest of cell-cycle progression, and induction of apoptosis. Importantly, the anti-WM effect exerted by NSC12 was also within the context of the supportive BM microenvironment, as demonstrated both in vitro and in vivo, using a disseminated humanized WM model. Taken together, our studies provide the evidence for an antitumor activity resulting from a specific targeting of the FG/FGFR axis, within the context of WM, thus representing a potential novel therapeutic strategy for WM patients.
Methods
Cells
A detailed description is provided within the supplemental Data, available on the Blood Web site.
Approval for these studies was obtained from the local ethics committee. Informed consent was obtained from all patients in accordance with the Declaration of Helsinki protocol.
Reagents
Pan-FGF trap molecule NSC12 was synthesized as described.27,28 A detailed description is provided within the supplemental Data.
MYD88-knock-in
Overexpression of MYD88 was obtained in BCWM1 and WMCL1 cells using precision LentiORF particles/MYD88(mGFP-tagged) and the corresponding empty/MOCK vector as control. Overexpression efficiency was performed by using quantitative reverse transcription polymerase chain reaction (qRT-PCR) and western blot.
Growth inhibition, viability, cell cycle, apoptosis, and mitosox assay
A detailed description is provided within the supplemental Data.
Adhesion assay
Adhesion of WM cells to primary WM BMSCs was evaluated by an in vitro adhesion assay, using Calcein AM-labeling of WM cells, with degree of fluorescence measured on a fluorescence plate reader (Varioskan LUX-ABS/FLUO Top/Bottom Lumi, Thermo Fisher Scientific, Waltham, MA), at excitation λ485 nm and emission λ520 nm, as described.29-31
Western blot analysis
WM cells were harvested and lysed using lysis buffer (Cell Signaling Technology, Beverly, MA) supplemented with 5 mM NaF, 2 mM Na3VO4, 1 mM phenylmethylsulfonyl fluoride, 5 μg/mL leupeptine, and 5 μg/mL aprotinin. Protein concentration in the supernatants was determined using the Bradford protein assay (Bio-Rad Laboratories). Whole-cell lysates were subjected to sodium dodecyl sulfate-polyacrylamide gel eletrophoresis and transferred to polyvinyldene fluoride membrane (Bio-Rad Laboratories, Hercules, CA). Immunoblotting was assessed using primary antibodies, as indicated within the supplemental Data.
Nuclear extraction and NF-κB activity
Nuclear extracts of the cells were prepared using the NE-PER Nuclear and Cytoplasmic Extraction Kit (Thermo Fisher Scientific) following the manufacturer's procedure, and as previously described.32,33
qRT-PCR analysis
For analysis of messenger RNA (mRNA) expression, total RNA was extracted using TRIzol Reagent (Invitrogen) according to manufacturer’s instructions, as previously reported.19,30 mRNA expression was analyzed by qRT-PCR using SYBR green dye for MYD88 and HCK, using a QS3 Real-Time PCR system (Life Technologies). All PCR reactions were run in triplicate. Ct values were normalized on glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and relative changes were calculated using the 2−ΔΔCt.
IL-6 enzyme-linked immunosorbent assay
Interleukin-6 (IL-6) secretion from primary WM bone marrow stromal cells (BMSCs) was assessed using the enzyme-linked immunosorbent assay (ELISA Kit for Interleukin-6; Cloud Clone Corp, Katy, TX) according to the manufacturer’s instructions.
A detailed description is provided within the supplemental Data.
Transcriptome profiling
RNA was extracted from WM cell lines (BCWM.1; MWCL.1) as previously reported.34,35 RNA samples were processed using WT PLUS Reagent Kit, according to manufacturer’s protocol (Thermo Fisher Scientific). Wide mRNA-transcriptome profiling was assessed using Clariom D Human array (Thermo Fisher Scientific).36-38
A detailed description is provided within the supplemental Data. Gene Set Enrichment Analysis (GSEA) was applied on global protein-coding gene expression profiles: significant gene sets were selected based on nominal P value < .05 and FDR < 0.25.34,35,39-41
In vivo studies
Systemic human xenograft experiments were performed according to the Italian laws (D.L. 116/92) that enforce the EU 86/109 Directive and were approved by the local animal ethics committee (Organismo Preposto al Benessere degli Animali, Università degli Studi di Brescia, Italy).
In vivo studies were performed as previously reported.2,35,42 A detailed description is provided within the supplemental Data.
Statistical analysis
Data are expressed as the mean ± standard error of the mean. Statistical analyses were performed by using GraphPad Prism 7.0 software. P values were generated using a 2-tailed Student t test to compare the differences between the 2 groups. A P value < .05 was considered statistically significant. GSEA:significant gene sets were selected based on nominal P value < .05 and FDR < 0.25.
Results
WM is characterized by activation of FGF/FGFR–induced signaling pathways: NSC12-dependent FGF/FGFR blockade affects WM cell survival
Previous studies have demonstrated the overexpression of FGFR3 in WM patients’ derived tumor cells4 ; nevertheless, a specific FGF/FGFR–targeting approach and the related functional sequelae have not been reported yet. Moreover, it is known how the FGF/FFGFR axis plays a crucial role in modulating the activation of prosurvival signaling pathways, including the PI3K-AKT, MAPK, and JAK-STAT cascades.43-45 We therefore interrogated the transcriptome signature of patients’ BM-derived CD19-positive cells and revealed a significant enrichment of PI3K-AKT, MAPK, and STAT3-related gene sets in WM cells as compared with their normal cellular counterpart, as shown by GSEA (Figure 1A). Of note, we are reporting on a significantly higher expression of FGFs (FGF2, FGF7, FGF12, FGF18) on primary WM cells as compared with their normal cellular counterpart (supplemental Figure 1A). Importantly, a peculiar phosphorylation level of FGFR3 was demonstrated using protein lysates of CD19+ cells obtained from the BM of WM patients (MYD88L265P mutated), as compared with CD19+ cells isolated from the BM and the peripheral blood of healthy individuals (supplemental Figure 2A).

NSC12 elicits antitumor activity in WM. (A) Enrichment plots of AKT-, STAT3, MAPK-related genes in primary WM patients’ derived CD19+ cells, as shown by performing GSEA (P < .05; FDR < 2 5%), using a publicly available data set (GEO6691). BCWM.1, MWCL.1, MEC.1 cells (B) or primary WM patients’ derived CD19+ cells (n. 3) (C), and normal PBMCs (D) were exposed to increasing concentrations of NSC12 (0.1 to 10 μM) for 48 hours. Cell survival was evaluated by MTT assay. Average of triplicate experiments ± standard deviation (SD) is shown. (E) BCWM.1 and MWCL.1 cells were exposed to NSC12 (6 μM) for 12 hours and subjected to wide transcriptome profiling, showing a significant inhibition of FGFR-signaling–related gene sets, as shown by performing GSEA (P < .05; FDR < 25%). (F) BCWM.1 and MWCL.1 cells were cultured in the presence or absence of NSC12 (0 to 6 μM; 6 hours). WM cells were then harvested, and cell lysates were subjected to western blot using anti-phoshpo(p)-FGFR3, -p-FSR2, -GAPDH. (G) BCWM.1 and MWCL.1 cells were cultured in the presence or absence of NSC12 (0 to 6 μM; 6 hours). WM cells were then harvested, and cell lysates were subjected to western blot using anti-p-AKT, -AKT, -p-GSK3β, -p-6SR, -p-ERK, -ERK, -p-JAK2, -p-STAT3, -GAPDH. MTT, 3-(4,5-dimethylthiazol-2-Yl)-2,5-diphenyltetrazolium bromide.
NSC12 elicits antitumor activity in WM. (A) Enrichment plots of AKT-, STAT3, MAPK-related genes in primary WM patients’ derived CD19+ cells, as shown by performing GSEA (P < .05; FDR < 2 5%), using a publicly available data set (GEO6691). BCWM.1, MWCL.1, MEC.1 cells (B) or primary WM patients’ derived CD19+ cells (n. 3) (C), and normal PBMCs (D) were exposed to increasing concentrations of NSC12 (0.1 to 10 μM) for 48 hours. Cell survival was evaluated by MTT assay. Average of triplicate experiments ± standard deviation (SD) is shown. (E) BCWM.1 and MWCL.1 cells were exposed to NSC12 (6 μM) for 12 hours and subjected to wide transcriptome profiling, showing a significant inhibition of FGFR-signaling–related gene sets, as shown by performing GSEA (P < .05; FDR < 25%). (F) BCWM.1 and MWCL.1 cells were cultured in the presence or absence of NSC12 (0 to 6 μM; 6 hours). WM cells were then harvested, and cell lysates were subjected to western blot using anti-phoshpo(p)-FGFR3, -p-FSR2, -GAPDH. (G) BCWM.1 and MWCL.1 cells were cultured in the presence or absence of NSC12 (0 to 6 μM; 6 hours). WM cells were then harvested, and cell lysates were subjected to western blot using anti-p-AKT, -AKT, -p-GSK3β, -p-6SR, -p-ERK, -ERK, -p-JAK2, -p-STAT3, -GAPDH. MTT, 3-(4,5-dimethylthiazol-2-Yl)-2,5-diphenyltetrazolium bromide.
On this basis, we next sought to interrogate the effect of the pan-FGF-trap, NSC12, WM cells (BCWM.1; MWCL.1); cells were cultured for 48 hours in the presence or absence of NSC12 (0 to 10 μM). NSC12 inhibited WM cell survival. NSC12 demonstrated similar activity on immunoglobulin M (IgM)-secreting cell lines (Figure 1B). We next evaluated and confirmed NSC12-induced anti-WM activity using primary BM-derived CD19+ cells isolated from patients with WM (Figure 1C). In contrast, NSC12 did not show cytotoxicity on peripheral blood mononuclear cell (PBMC)-derived CD19+ cells isolated from healthy volunteers (Figure 1D). Taken together, these results demonstrate that NSC12 triggers significant cytotoxicity in tumor cell lines and patient CD19+ WM cells, without affecting normal PBMCs.
Given the crucial role of CXCR4C1013G in supporting WM pathogenesis, and its occurrence in ∼30% of the cases,13,19,21,46 we have evaluated the activity of NSC12 using CXCR4C1013G-mutated BCWM.1 and MWCL.1 and confirmed the NSC12-dependent anti-WM activity also within the context of CXCR4C1013G-mutated BCWM.1 and MWCL.1. Importantly, the observed phenotype was supported by inhibition of p-FGF3 in CXCR4C1013G-mutated BCWM.1 and MWCL.1 exposed to NSC12 (supplemental Figure 3A-B). We next tested the effect of pan-FGF trap NSC12 on targeting the activation of FGF/FGFR–induced signaling in WM cells, at both mRNA and protein level. By performing wide-transcriptome profiling of NSC12-treated WM cells, we first demonstrated an efficacious silencing of FGFR-activated downstream signaling, in both BCWM.1 and MWCL.1 (Figure 1E). Importantly, we confirmed the ability of NCS12 to inhibit phosphorylation of both FGFR3 and FGFR-substrate2 (FRS2) (Figure 1F); supported by downregulation of prosurvival pathways, including phospho-AKT, with the downstream phospho-GSK3β and -S6R; phospho-JAK2 and -STAT3; and phospho-ERK, as demonstrated in both BCWM.1 and MWCL.1 (Figure 1G).
To gain further insights into the potential molecular mechanisms underlying the NSC12-dependent anti-WM activity, we compared NSC12-treated vs untreated WM cell lines and identified significant differences in their transcriptional profiles associated with the modulation of several biological processes involved in the regulation of cell-cycle progression, DNA replication, and cell division, thus suggesting a role of NSC12 in targeting WM cell growth (Figure 2A; supplemental Table 1). We next investigated whether the observed NSC12-induced transcriptome changes were, indeed, linked to a functional phenotype in WM cells: NSC12-treated BCWM.1 and WMCL.1 cells presented with a significant cell-cycle arrest, as documented by S-phase reduction, coupled with an increase in sub-G0 phase, thus suggesting a proapoptotic effect (Figure 2B). This prompted us to dissect the ability of NSC12 to enhance apoptosis in WM cells and demonstrated an NCS12-dependent induction of the apoptotic phenotype, together with reduction of the % of WM viable cells, as evidenced by annexin/PI staining and flow cytometry analysis (Figure 2C). We next defined mechanisms whereby NCS12 induces apoptosis in WM and demonstrated that NSC12 enhances caspase-8 and PARP-cleavage (Figure 2D). In order to investigate potential mechanisms supporting the NSC12-induced apoptotic phenotype, we looked into the transcriptome changes occurring in NSC12, showing induction of oxidative stress in NSC12-treated WM cells (Figure 2E). To further demonstrate the functional sequelae, we next investigated whether NSC12 could affect mitochondrial reactive oxygen species (mtROS) and demonstrated an increase of mtROS in NSC12-treated WM cells (Figure 2F). Of note, this was supported by enhanced DNA damage as shown by upregulation of p-H2AX, and oxidative stress-protective transcription factor NRF2 downregulation (Figure 2G).
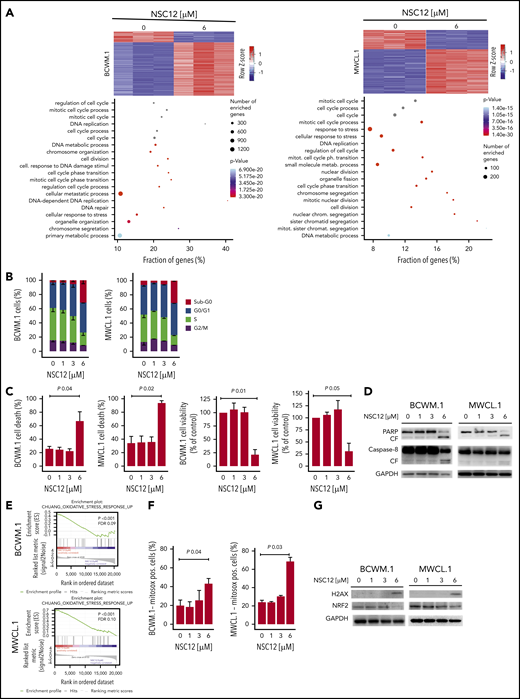
NSC12 modulates the transcriptome signature in WM cells, resulting in antiproliferative and proapoptotic phenotype. (A) Heatmaps of differentially expressed protein-coding transcripts in BCWM.1 (325 upregulated, 1586 downregulated) and MWCL.1 cells (269 upregulated, 614 downregulated). Blue-red color scale was used to set rows with mean zero and SD one. Genes selected at q value 0 and fold change >2. Plot of the 20 most significant differentially expressed protein coding gene lists enriched in BCWM.1 and MWCL.1 (GO-BP terms). (B) Cytofluorimetric analysis of cell cycle performed using BCWM.1 and MWCL.1 exposed to NSC12 (0 to 6 μM; 12 hours). Average of triplicate experiments ± SD is shown. (C) Annexin V/PI staining was performed using BCWM.1 and MWCL.1 cells exposed to NSC12 (0 to 6 μM; 24 hours). Percent of both dead cells and viable cells is shown. Average of triplicate experiments ± SD is shown. (D) BCWM.1 and MWCL.1 cells were cultured in the presence or absence of NSC12 (0 to 6 μM; 24 hours). WM cells were then harvested, and cell lysates were subjected to western blot using anti-PARP, -Caspase 8, and -GAPDH antibodies. (E) BCWM.1 and MWCL.1 cells were exposed to NSC12 (6 μM) for 12 hours and subjected to wide transcriptome profiling, showing a significant inhibition of oxidative stress response-related gene sets, as shown by performing GSEA (P < .05; FDR < 25%). (F) BCWM.1 and MWCL.1 cells were treated with NSC12 (0 to 6 μM) for 12 hours and subjected to cytofluorimetric analysis of mtROS production (Mitosox). (G) BCWM.1 and MWCL.1 cells were cultured in the presence or absence of NSC12 (0 to 6 μM; 12 hours). WM cells were then harvested, and cell lysates were subjected to western blot using anti-H2AX, -NRF2, and -GAPDH antibodies.
NSC12 modulates the transcriptome signature in WM cells, resulting in antiproliferative and proapoptotic phenotype. (A) Heatmaps of differentially expressed protein-coding transcripts in BCWM.1 (325 upregulated, 1586 downregulated) and MWCL.1 cells (269 upregulated, 614 downregulated). Blue-red color scale was used to set rows with mean zero and SD one. Genes selected at q value 0 and fold change >2. Plot of the 20 most significant differentially expressed protein coding gene lists enriched in BCWM.1 and MWCL.1 (GO-BP terms). (B) Cytofluorimetric analysis of cell cycle performed using BCWM.1 and MWCL.1 exposed to NSC12 (0 to 6 μM; 12 hours). Average of triplicate experiments ± SD is shown. (C) Annexin V/PI staining was performed using BCWM.1 and MWCL.1 cells exposed to NSC12 (0 to 6 μM; 24 hours). Percent of both dead cells and viable cells is shown. Average of triplicate experiments ± SD is shown. (D) BCWM.1 and MWCL.1 cells were cultured in the presence or absence of NSC12 (0 to 6 μM; 24 hours). WM cells were then harvested, and cell lysates were subjected to western blot using anti-PARP, -Caspase 8, and -GAPDH antibodies. (E) BCWM.1 and MWCL.1 cells were exposed to NSC12 (6 μM) for 12 hours and subjected to wide transcriptome profiling, showing a significant inhibition of oxidative stress response-related gene sets, as shown by performing GSEA (P < .05; FDR < 25%). (F) BCWM.1 and MWCL.1 cells were treated with NSC12 (0 to 6 μM) for 12 hours and subjected to cytofluorimetric analysis of mtROS production (Mitosox). (G) BCWM.1 and MWCL.1 cells were cultured in the presence or absence of NSC12 (0 to 6 μM; 12 hours). WM cells were then harvested, and cell lysates were subjected to western blot using anti-H2AX, -NRF2, and -GAPDH antibodies.
NSC12-mediated inhibition of FGFR-signaling targets WM cells by silencing MYD88 and MYD88-driven pathways
NSC12 induced a significant silencing of FGFR-related gene sets in WM cells: indeed, NSC12-dependent inhibition of PI3K-AKT- and STAT3-signaling related genes was demonstrated in both BCWM.1- and MWCL.1-treated cells as compared with the untreated WM cells, as shown by GSEA (Figure 3A; supplemental Figure 4A). In addition, the MAPK cascade was shown to be silenced in NSC12-treated WM cells, as indicated by a significant reduction of MAPK signaling cascade-related gene sets in BCWM.1 and MWCL1 cells (supplemental Figure 4B). Of note, among the silenced genes representing the PI3/AKT/mTOR signaling gene set, we discovered MYD88 being significantly downregulated in NSC12-treated BCWM.1 and MWCL.1 cells (Figure 3A). These findings were also corroborated by showing the NSC12-dependent downregulation of MYD88-related genes in both BCWM.1 and MWCL.1, as compared with the relative untreated controls. We next confirmed that MYD88 protein expression was also silenced in WM cells exposed to NSC12, followed by inhibition of MYD88-driven pathways, such as BTK-, SYK-, and IRAK1-phosphorylation (Figure 3B). We next interrogated whether NSC12 could modulate the expression of wild-type MYD88 and the L265P-MYD88 mutated gene and described an efficacious NSC12-dependent silencing of both wild-type and mutated MYD88 gene (supplemental Figure 5). It has been reported that mutated MYD88 triggers transcription of hematopoietic cell kinase (HCK), known to act as a prosurvival factor in WM,and aberrantly upregulated in WM cells.47,48 To further look into the functional relevance of NSC12 to halt not only MYD88 but also HCK, we investigated the effects exerted by NSC12 in modulating HCK expression and found a significant silencing of HCK transcript in WM cells exposed to NSC12 (Figure 3C). Overall, these findings provide novel insights into the ability of NSC12 to target MYD88 in WM cells.
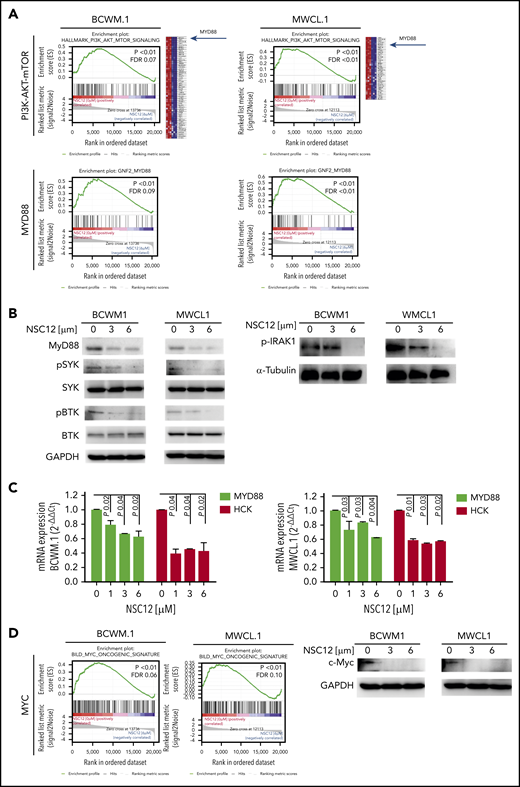
NSC12 mediates inhibition of MYD88 in WM cells. (A) BCWM.1 and MWCL.1 cells were exposed to NSC12 (6 μM) for 12 hours and subjected to wide transcriptome profiling, showing a significant inhibition of PI3K-AKT-mTOR- and MYD88-related gene sets, as shown by performing GSEA (P < .05; FDR < 25%). (B) BCWM.1 and MWCL.1 cells were cultured in the presence or absence of NSC12 (0 to 6 μM; 6 hours). WM cells were then harvested, and cell lysates were subjected to western blot using anti-MYD88, -p-SYK, -SYK, -p-BTK, -BTK, and -GAPDH antibodies. (C) BCWM.1 and MWCL.1 cells were treated with NSC12 (0 to 6 μM; 6 hours), subjected to RNA extraction, and evaluated for MYD88 and HCK mRNA levels by using qRT-PCR (2−ΔΔCt), with normalization to GAPDH (P, P values). (D) BCWM.1 and MWCL.1 cells were cultured in the presence or absence of NSC12 (0 to 6 μM; 6 hours). WM cells were then harvested, and cell lysates were subjected to western blot using anti-c-Myc and -GAPDH antibodies.
NSC12 mediates inhibition of MYD88 in WM cells. (A) BCWM.1 and MWCL.1 cells were exposed to NSC12 (6 μM) for 12 hours and subjected to wide transcriptome profiling, showing a significant inhibition of PI3K-AKT-mTOR- and MYD88-related gene sets, as shown by performing GSEA (P < .05; FDR < 25%). (B) BCWM.1 and MWCL.1 cells were cultured in the presence or absence of NSC12 (0 to 6 μM; 6 hours). WM cells were then harvested, and cell lysates were subjected to western blot using anti-MYD88, -p-SYK, -SYK, -p-BTK, -BTK, and -GAPDH antibodies. (C) BCWM.1 and MWCL.1 cells were treated with NSC12 (0 to 6 μM; 6 hours), subjected to RNA extraction, and evaluated for MYD88 and HCK mRNA levels by using qRT-PCR (2−ΔΔCt), with normalization to GAPDH (P, P values). (D) BCWM.1 and MWCL.1 cells were cultured in the presence or absence of NSC12 (0 to 6 μM; 6 hours). WM cells were then harvested, and cell lysates were subjected to western blot using anti-c-Myc and -GAPDH antibodies.
We next performed rescue experiments by overexpressing MYD88 in WM cells and tested the ability of NSC12 to target MYD88-overepxressing WM cells. Efficacy of knock-in experiments was first confirmed at both transcriptome and protein level, in both BCWM.1- and MWCL.1-engineered cells (supplemental Figure 6A-B). The forced MYD88 expression in WM cells significantly impaired NSC12-induced cell death, thus further confirming the ability of NSC12 to target WM cells via MYD88 (supplemental Figure 6C).
MYD88 deficiency has been linked to a significant decrease of c-Myc.49 Given the oncogenic role of Myc described also in hematologic malignancies,50-52 we next investigated the functional consequences of NSC12-dependent MYD88 silencing in modulating Myc in WM cells and demonstrated a significant downregulation of Myc at both transcriptome and protein level (Figure 3D; supplemental Figure 7A).
Previous studies have shown how MYD88 may promote NF-κB pathway activation,15 thus resulting in modulation of growth and survival of WM cells. We therefore sought to dissect the effect of NSC12 in affecting the NF-κB pathway. We first confirmed the ability of NSC12 to inhibit NF-κB–related gene sets, as shown by GSEA in both BCWM.1 and MWCL.1 (Figure 4A). To further corroborate the functional relevance of NSC12-dependent modulation of the NF-κB in WM cells, we investigated the effect of NSC12 on the NF-κB–p65 DNA-binding activity, studying nuclear extracts from NSC12-treated WM cells. We showed that tumor necrosis factor-α (TNF-α)–induced NF-κB–p65 recruitment to the nucleus in BCWM.1 and MWCL.1 cells was significantly inhibited by NCS12 (Figure 4B). In order to investigate the effect of NSC12 in modulating NF-κB pathway at the protein level, we first investigated the status of inhibitory protein IκB in WM cells exposed to NSC12, in the presence or absence of TNF-α, demonstrating an NSC12-dependent upregulation of phospho(p)-IκB, which resulted in total IκB degradation (Figure 4C). In parallel, immunoblotting from nuclear extracts demonstrated that TNF-α–induced p65 phosphorylation and p50NF-κB expression were inhibited by NCS12. We next examined whether the NCS12 also altered the noncanonical NF-κB pathway. Immunoblotting from nuclear extracts showed that NCS12 inhibited the expression of p52 and RelB, which are activated mostly through the noncanonical pathway (Figure 4D). Taken together, these data demonstrate that NCS12 targets both the canonical and the noncanonical NF-κB pathways in WM cells.
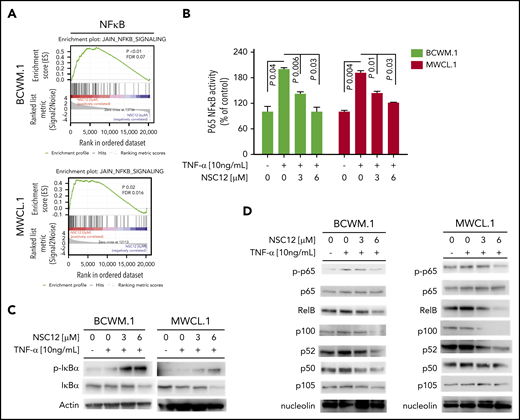
NSC12 targets WM cells by inhibiting NF-κB activity. (A) BCWM.1 and MWCL.1 cells were exposed to NSC12 (6 μM) for 12 hours and subjected to wide transcriptome profiling, showing a significant inhibition of NF-κB signaling-related gene sets, as shown by performing GSEA (P < .05; FDR < 25%). (B) BCWM.1 and MWCL.1 cells were cultured with NSC12 (0 to 6 μM), for 4 hours, and then exposed TNF-α (10 ng/mL) was added for the last 20 minutes. NF-κBp65 transcription factor binding to its consensus sequence on the plate-bound oligonucleotide was studied from nuclear extracts. Wild-type and mutant are wild-type and mutated consensus competitor oligonucleotides, respectively. All results represent means (± SD) of triplicate experiments. (C) BCWM.1 and MWCL.1 cells were cultured in the presence or absence of NSC12 (0 to 6 μM), for 4 hours, and then exposed TNF-α (10 ng/mL) was added for the last 20 minutes. WM cells were then harvested, and cell lysates were subjected to western blot using anti-p-IkBa, -IkBa, and -actin antibodies. (D) BCWM.1 and MWCL.1 cells were cultured in the presence or absence of NSC12 (0 to 6 μM), for 4 hours, and then exposed TNF-α (10 ng/mL) was added for the last 20 minutes. WM cells were then harvested, and nuclear protein lysates were extracted and subjected to western blot using anti-p-p65, -p65, -RelB, -p100, -p52, -p50, -p105, and -nucleolin antibodies.
NSC12 targets WM cells by inhibiting NF-κB activity. (A) BCWM.1 and MWCL.1 cells were exposed to NSC12 (6 μM) for 12 hours and subjected to wide transcriptome profiling, showing a significant inhibition of NF-κB signaling-related gene sets, as shown by performing GSEA (P < .05; FDR < 25%). (B) BCWM.1 and MWCL.1 cells were cultured with NSC12 (0 to 6 μM), for 4 hours, and then exposed TNF-α (10 ng/mL) was added for the last 20 minutes. NF-κBp65 transcription factor binding to its consensus sequence on the plate-bound oligonucleotide was studied from nuclear extracts. Wild-type and mutant are wild-type and mutated consensus competitor oligonucleotides, respectively. All results represent means (± SD) of triplicate experiments. (C) BCWM.1 and MWCL.1 cells were cultured in the presence or absence of NSC12 (0 to 6 μM), for 4 hours, and then exposed TNF-α (10 ng/mL) was added for the last 20 minutes. WM cells were then harvested, and cell lysates were subjected to western blot using anti-p-IkBa, -IkBa, and -actin antibodies. (D) BCWM.1 and MWCL.1 cells were cultured in the presence or absence of NSC12 (0 to 6 μM), for 4 hours, and then exposed TNF-α (10 ng/mL) was added for the last 20 minutes. WM cells were then harvested, and nuclear protein lysates were extracted and subjected to western blot using anti-p-p65, -p65, -RelB, -p100, -p52, -p50, -p105, and -nucleolin antibodies.
NSC12-dependent anti-WM activity is enhanced when used in combination with other anti-WM agents
By interrogating the transcriptome profile of NSC12-treated cells, we found a significant inhibition of ubiquitin proteasome degradation-related genes (Figure 5A). Therefore, the NSC12-dependent inhibition of the ubiquitin proteasome degradation could result in the enhancement of the polyubiquitinated protein cargo in WM cells. This hypothesis prompted us to investigate any potential impact on modulation of endoplasmic reticulum (ER) stress and unfolded protein response (UPR), known to be activated by the accumulation of misfolded proteins within the ER: we found a significant enrichment of ER-related genes in NSC12-treated BCWM.1 and MWLC.1 cells (Figure 5B), together with an enhanced initiation of the UPR (Figure 5C). We therefore investigated whether these changes could lead to an enhanced antitumor activity of NSC12 in the presence of proteasome inhibitors (PIs) and found how the combined use of either bortezomib or carfilzomib with NSC12 resulted in a more significant dose-dependent inhibition of WM cell survival (Figure 5D; supplemental Figure 8A; P < .05). By adding PIs, we may further enhance ER stress and UPR within WM cells, thus explaining, at least in part, the combinatory effect of NSC12 used with PIs, bortezomib and carfilzomib. Similarly, given the ability of NSC12 itself to inhibit mTOR signaling-related genes (Figure 3A), and to target and silence MYD88 and MYD88-driven pathways, such as BTK, the combinatory treatment of NSC12 with either mTOR- or BTK-inhibitors was evaluated, showing a more significant dose-dependent inhibition of WM cell survival (supplemental Figure 8B-C; P < .05).

NSC12 leads to enhanced anti-WM activity when used in combination with PIs. (A-C) BCWM.1 and MWCL.1 cells were exposed to NSC12 (6 μM) for 12 hours and subjected to wide transcriptome profiling, showing a significant inhibition of antigen processing ubiquitination proteasome degradation- (A), ER stress- (B), and UPR (C) -related gene sets, as shown by performing GSEA (P < .05; FDR < 25%). (D) BCWM.1 and MWCL.1 cells were treated with NSC12 (0.3 to 3 μM) or bortezomib (2.5 to 5 nM) as single agents, or the combination. Modulation of cell survival at 48 hours was tested on WM cells using MTS assay. Average of triplicate experiments ± SD is shown. P, P value.
NSC12 leads to enhanced anti-WM activity when used in combination with PIs. (A-C) BCWM.1 and MWCL.1 cells were exposed to NSC12 (6 μM) for 12 hours and subjected to wide transcriptome profiling, showing a significant inhibition of antigen processing ubiquitination proteasome degradation- (A), ER stress- (B), and UPR (C) -related gene sets, as shown by performing GSEA (P < .05; FDR < 25%). (D) BCWM.1 and MWCL.1 cells were treated with NSC12 (0.3 to 3 μM) or bortezomib (2.5 to 5 nM) as single agents, or the combination. Modulation of cell survival at 48 hours was tested on WM cells using MTS assay. Average of triplicate experiments ± SD is shown. P, P value.
NSC12-dependent FGF/FGFR blockade affects WM cells in the context of BM microenvironment, leading to antitumor activity in vivo
It has been widely demonstrated that BM microenvironment confers growth and induces drug resistance within the setting of B-cell malignancies, including WM.29,30,53,54 We therefore sought to evaluate the antitumor activity of NSC12 against WM cells in the context of the BM milieu. BCWM.1 cells were cultured with NSC12 in the presence or absence of primary WM BMSCs for 48 hours. Adherence of BCWM.1 cells to BMSCs triggered a significant increase in the proliferation, which was inhibited by NSC12 in a dose-dependent manner (Figure 6A). Similar findings were obtained using MWCL.1 as well as other and IgM-secreting low-grade lymphoma cells (supplemental Figure 9A-B). We next confirmed the efficacy of NSC12 in silencing BMSC-induced FGFR3 phosphorylation, paralleled by silencing of prosurvival pathways, including phospho-AKT, with the downstream phospho-GSK3β, phospho-ERK and -STAT3 (Figure 6B). Moreover, BMSC-induced upregulation of adhesion-related proteins, phospho-SRC, and -cofilin was also inhibited in WM cells exposed to NSC12 (Figure 6B). Considering the relevance of SRC and cofilin in supporting cell adhesion, we next evaluated the functional sequelae of NSC12 in affecting WM cell adhesion and demonstrated the NSC12-dependent inhibition of both WM and IgM-secreting low-grade lymphoma cells to primary WM BMSCs (Figure 6C). Overall, these findings confirm the ability of NSC12 to target WM cells even within the context of the supportive BM niche.

NSC12 targets WM cells within the context of the BM microenvironment both in vitro and in vivo. (A) Modulation of cell proliferation of BCWM.1 cells exposed to NSC12 (0 to 6 μM; 48 hours) cocultured in the presence or absence of WM patient-derived BMSCs was assessed by CellTiter-Glo. (B) BCWM.1 and MWCL.1 cells were cultured in the presence or absence of WM patient’s derived BMSC and exposed to NSC12 (0 to 6 μM; 6 hours). WM cells were then harvested, and cell lysates were subjected to western blotting, with the use of antibodies anti-p-FGFR3, -p-AKT, -p-GSK3β, -p-.ERK, p-STAT3, -p-Cofilin, p-SRC, and -GAPDH (C) Adhesion of BCWM.1, MWCL.1, MEC.1, WMWSU cells to WM patient-derived BMSCs exposed to NSC12 (0 to 6 μM; 4 hours) was evaluated by an in vitro adhesion assay, using Calcein AM labeling of WM cells, with degree of fluorescence measured on a spectrophotometer (λ485 nm to λ520 nm). (D-E) SCID/Bg mice were injected with BCWM.1-mCherry+/Luc+ cells and treated with either NSC12 (7.5 mg/kg, every other day) or vehicle control. Detection of tumor growth was performed by measuring bioluminescence imaging (BLI) intensity at different time points post-WM cell injection (days 7, 10, 14). P, P value. (F) WM cell BM colonization was evaluated ex vivo from harvested femurs, using immunostaining for human (h)-CD20. H.E., hematoxylin-eosin staining (×20). (G) BM mononuclear cells were harvested ex vivo from femurs, subjected to RNA extraction, and evaluated for MYD88, HCK mRNA levels by using qRT-PCR (2−ΔΔCt), with normalization to GAPDH (*P < .05).
NSC12 targets WM cells within the context of the BM microenvironment both in vitro and in vivo. (A) Modulation of cell proliferation of BCWM.1 cells exposed to NSC12 (0 to 6 μM; 48 hours) cocultured in the presence or absence of WM patient-derived BMSCs was assessed by CellTiter-Glo. (B) BCWM.1 and MWCL.1 cells were cultured in the presence or absence of WM patient’s derived BMSC and exposed to NSC12 (0 to 6 μM; 6 hours). WM cells were then harvested, and cell lysates were subjected to western blotting, with the use of antibodies anti-p-FGFR3, -p-AKT, -p-GSK3β, -p-.ERK, p-STAT3, -p-Cofilin, p-SRC, and -GAPDH (C) Adhesion of BCWM.1, MWCL.1, MEC.1, WMWSU cells to WM patient-derived BMSCs exposed to NSC12 (0 to 6 μM; 4 hours) was evaluated by an in vitro adhesion assay, using Calcein AM labeling of WM cells, with degree of fluorescence measured on a spectrophotometer (λ485 nm to λ520 nm). (D-E) SCID/Bg mice were injected with BCWM.1-mCherry+/Luc+ cells and treated with either NSC12 (7.5 mg/kg, every other day) or vehicle control. Detection of tumor growth was performed by measuring bioluminescence imaging (BLI) intensity at different time points post-WM cell injection (days 7, 10, 14). P, P value. (F) WM cell BM colonization was evaluated ex vivo from harvested femurs, using immunostaining for human (h)-CD20. H.E., hematoxylin-eosin staining (×20). (G) BM mononuclear cells were harvested ex vivo from femurs, subjected to RNA extraction, and evaluated for MYD88, HCK mRNA levels by using qRT-PCR (2−ΔΔCt), with normalization to GAPDH (*P < .05).
Given the ability of BMSCs to release IL-6 upon stimulation of FGF, and considering the relevance of IL-6 as a well-known cytokine that enhances cell proliferation within the context of B-cell lymphoproliferative disorders, including WM,55-59 we next investigated the effect of NSC12 to potentially alter the IL-6 secretion from the WM BM niche. We therefore exposed primary WM-BMSCs to increasing concentration of NSC12 and investigated whether NSC12 treatment could modulate IL-6 secretion, by evaluating the conditioned medium of NSC1212-treated BMSCs, as compared with control. Our findings demonstrate the efficacy of NSC12 to halt secretion of IL-6 from WM-BMSCs, thus further corroborating the biological relevance of NSC12 within the context of WM (supplemental Figure 9C).
The impact of NCS12 on WM tumor growth in vivo was evaluated using a disseminated humanized WM model.19,42 BCWM.1 M-Cherry-Luc tumor-bearing mice were treated with either NSC12 or vehicle control. NSC12-treated mice presented with a significant inhibition of WM tumor growth as demonstrated by bioluminescence imaging analysis (Figure 6D-E). Moreover, NSC12-dependent reduction of WM tumor cell infiltration of the BM was confirmed by immunohistochemistry performed on harvested femurs, in comparison with vehicle-treated mice (Figure 6F). To further confirm the in vitro findings suggesting a role of NSC12 in silencing MYD88 in WM cells, we next assessed MYD88-mRNA expression using harvested BM femurs and found a significant reduction of MYD88 in NSC12-treated mice (Figure 6G). As a further confirmation of the in vivo NSC12-dependent inhibition of MYD88, we demonstrated a significant inhibition of the MYD88-downstream target HCK (Figure 6G).
Discussion
We are reporting on the antitumor activity exerted by NSC12, a novel pan-FGF trap, responsible for specific FGF/FGFR axis blockade, within the context of WM. Previous studies have demonstrated the overexpression of FGFR3 in WM patients’ derived tumor cells, as compared with their normal cellular counterpart,4 thus indicating FGFR3 as a potential therapeutic target in WM. We are now showing an enhanced expression of different FGFs and FGFR3 phosphorylation in WM cells, thus suggesting the existence of a reinforced FGF/FGFR3 signaling in WM, despite the lack of a peculiar t(4;14) in WM tumor cells. We acknowledge WM patients do not present with the t(4;14), as described in multiple myeloma (MM), where overexpression of FGFR3 protein occurs in ∼70% of patients with the t(4;14) translocation; of note, 25% of the t(4,14)-positive MM cases do not express FGFR3.60-62 Additional mechanisms by which FGFR3 is dysregulated independently from the presence of t(4;14) could occur; indeed, enhanced FGFR3 expression has been reported in t(4;14)-negative MM, and the same phenotype has been also demonstrated within the context of myeloid malignancies, without a correlation with chromosomal abnormalities.63-65 For instance, FGFR3 overexpression, and its oncogenic role, has been reported in t(4;14)-negative chronic myelogenous leukemia cells.65 We may therefore hypothesize how the FGFR3 overexpression may not be just an epiphenomenon in t(4;14) MM, but an important part of the malignant phenotype, independent of the t(4;14), as shown in both lymphoid and myeloid tumors.63-65 Within the specific context of WM, an autocrine FGF stimulation could support the reinforced expression of FGFR3. However, the existence of other mechanisms cannot be excluded: for instance, our studies are not considering potential epigenetic modifications, or a BM milieu-WM cell cross-talk that could lead to activation of the FGF/FGFR signaling. Further studies are therefore needed to address these hypotheses.
Dovitnib-dependent multi-RTK inhibition has been used, aiming to neutralize FGFR3 in WM4 ; nevertheless, this strategy will necessarily lead to inhibition of several other RTKs, including, among others, vascular endothelial growth factor receptor, platelet-derived growth factor receptor, FLT3, stem cell factor receptor, colony-stimulating factor receptor 1, thus contributing to toxicities due to off-target effects, such as cardiovascular, gastrointestinal, and metabolic toxicities.
Moreover, by evaluating the pan-FGF trap in WM, we are able to use NSC12 as a tool to interrogate the molecular changes occurring in WM, resulting from NSC12-dependent FGF/FGFR blockade.
More recently, the extracellular FGF interactome has allowed the characterization of a variety of natural macromolecular binders able to interact with different members of the FGF family, thus modulating their bioavailability and biological activity. The identification of FGF binding domains has led to the development of molecules acting as extracellular FGF traps. This is the case of the pentraxin-3–derived small molecule NSC12.28
Differently from TK-FGFR inhibitors, FGF traps were reported not to induce signs of systemic toxicity in animals models28,66 In particular, even though potential NSC12-dependent off-targets effects cannot be completely excluded, several experiments have already been conducted, demonstrating the FGF/FGFR specificity of NSC12.28 Indeed, FGF/FGFR-insensitive cells do not respond to NSC12; FGF/FGFR-dependent cells lose the sensitivity to NSC12 after transduction with mutant/constitutively activated FGFR, and moreover, an excess of FGF2 abolishes the effect of NSC12.28
In our system, NSC12-treated WM cells presented with inhibition of tumor cell proliferation, cell-cycle arrest, and induction of apoptosis through caspase-dependent mechanisms. Of note, NSC12 was confirmed to target WM cells even in the presence of the BM microenvironment, known to support WM tumor growth and to mediate drug resistance.29,30,53,54 Of note, CXRC4C1013G WM cells were shown to be equally sensitive to NSC12 treatment.
We performed wide-transcriptome profiling of NSC12-treated WM cells, which allowed us to first confirm blockade of the FGFR-signaling, and, most importantly, to provide novel insights into the potential molecular mechanisms underlying the observed NSC12-dependent antitumor effect. Interestingly, we are reporting, for the first time, on the efficacious silencing of MYD88 in WM cells, as a result of the NSC12-induced FGF/FGFR-blockade, followed by inhibition of MYD88-driven pathways, such as BTK- and SYK-phosphorylation, as well as the MYD88-downstream target HCK.47,48 As a further demonstration of the functional impact of NSC12-dependent targeting of MYD88, we could confirm the inhibition of both canonical and noncanonical NF-κB in NSC12-treated WM cells.
It has been shown how MYD88-ERK signalling may contribute to enhanced tumor growth in colon cancer models, as a result of increased c-Myc protein stability.49 Indeed, ERK activation leads to c-Myc phosphorylation, which prevents its ubiquitination and the subsequent c-Myc proteasome-mediated degradation.49 Our studies are now reporting NSC12-dependent inhibition of MYD88, coupled with silencing of the MAPK-ERK signaling cascade, which may therefore explain, at least in part, the NSC12-induced Myc-silencing in WM cells. An additional explanation may reflect previous studies reporting on the cooperation of FGFR3 and Myc in supporting the progression of B-cell lymphoid malignancies.50 Similarly, recent evidence has demonstrated how FGF trapping is responsible for inhibition of MM tumor growth via c-Myc degradation-induced mitochondrial oxidative stress.2 Overall, NSC12-dependent FGF trapping may result in Myc-silencing, because of an indirect effect, mediated by NSC12-induced MYD88 silencing; or a direct effect, because of its own FGF-trapping activity responsible for FGF/FGFR axis blockade.
Of note, the activity of NSC12 was tested in combination with other anti-WM agents, such as PIs, including both carfilzomib and bortezomib. Our data suggest how the combination of NSC12 and PIs induced a significantly higher anti-WM activity, as compared with each drug used as monotherapy. These data seem to be specific for the WM context, as previous studies have not reported a similar behavior within the context of MM, where the combination of NSC12 and PIs showed a limited therapeutic window.2 This could be explained, at least in part, by the presence of NSC12-induced transcriptome changes in WM cells, demonstrating a downregulation of genes related to ubiquitination and proteasome degradation, responsible for enhanced ER stress and UPR: by reducing proteasome degradation, NSC12 could favor the polyubiquitinated protein cargo in WM cells, and by adding PIs, the combinatory regimen could further enhance antigen polyubiquitination, thus ultimately result higher WM cell toxicity.
A disseminated humanized WM in vivo model was adopted to confirm the antineoplastic activity exerted by NSC12, showing inhibited BM WM tumor cell infiltration. NSC12 induced Myd88 silencing, as validated on harvested BM, ex vivo.
Overall, our studies are reporting on the use of NSC12, as a novel potential therapeutic strategy to specifically halt the FGF/FGFR axis in WM and demonstrate how the observed anti-WM activity exerted by NSC12 may be driven, at least in part, by inhibition of MYD88.
Files related to transcriptome profiling of NSC12-treated BCWM.1 and MWCL.1 cells are available from the Gene Expression Omnibus (GEO), accession number GSE155155. Files related to gene expression profiling of BM patients’ derived WM cells are available from accession numbers GEO9656 and GEO6691.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
This work is supported by Fondazione AIRC, Fondazione Regionale per la Ricerca (A.M.R.), Biomedica, ERA-NET TRANSCAN-2: Associazione Italiana Contro le Leucemie-linfomi e Mieloma (AIL) Brescia, AIRC IG 16722, Italian Ministry of Health (Ricerca Corrente 2019) (A.N.), Fondazione Cariplo grant 2016-0570 (A.G.), Associazione Italiana Ricerca sul Cancro (AIRC IG 2019-ID.23151) (R.R.), and a fellowship by Fondazione Veronesi (F.M.).
Authorship
Contribution: A.M.R. and M.P. conceived and designed the study; A.M.R. and A.S. wrote the manuscript; A.M.R. and M.P. supervised the study; A.S., A.G., and C. Caprio performed the in vitro studies; A.A., M. Motta, D.R., G.R., C. Cattaneo, and A.T. provided patient samples; C.F., R.R., and F.M. performed the xenograft disseminated in vivo studies; A.G. performed the IHC studies; A.M.R., A.N., and M.P. reviewed the manuscript; K.T. and V.F. performed the transcriptome profiling; and R.C., N.B., and M. Mor conceived and performed the NSC12 synthesis.
Conflict-of-interest disclosure: A.M.R. has received research funding from AstraZeneca, European Hematology Association, Transcan2-ERANET, and Italian Association for Cancer Research (Fondazione AIRC) and has received honoraria from Amgen, Celgene, and Janssen. G.R. has received consultancy fees from Daiichi-Sankyo; served on advisory boards for Roche, Janssen, Celgene, Amgen, Gilead, Sanofi, Abbvie, Pfizer, Jazz, and Astellas; and has received honoraria for lectures for Novartis, Mundipharma, BMS, and Sandoz. The remaining authors declare no competing financial interests.
Correspondence: Aldo M. Roccaro, ASST Spedali Civili di Brescia, Clinical Research Development and Phase I Unit, P.le Spedali Civili di Brescia, n. 1, 25123, Brescia, BS, Italy; e-mail: aldomaria.roccaro@asst-spedalicivili.it; and Marco Presta, Department of Molecular and Translational Medicine, University of Brescia, V.le Europa 11, 23123, Brescia, BS, Italy; e-mail: marco.presta@unibs.it.