

In this issue of Blood, 1 reveal the first detailed structure of the key hemostatic complex formed between factor VIII (FVIII) and von Willebrand factor (VWF).
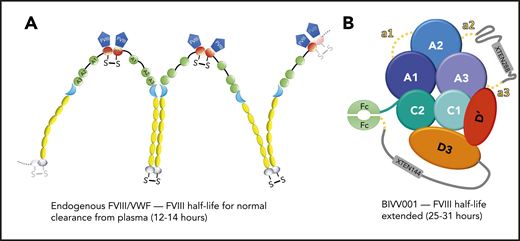
Endogenous VWF binding to FVIII vs bioengineered BIVV001. (A) Endogenous VWF complexed with FVIII is cleared from plasma with a half-life of 12 to 14 hours. FVIII (blue) binds to VWF via the D′D3 domain (red). VWF multimers are linked by disulfide bonds at the N-terminal D′D3 as well as the C-terminal CK domain (gray). The VWF-A domains (green) bear binding sites for platelet GP Ibα (A1), ADAMTS13 cleavage site (A2), and collagen (A1 and A3). (B) Bioengineered recombinant FVIII (rFVIII)/VWF-D′D3 construct, namely BIVV001, has an extended half-life of 25 to 31 hours, which is a threefold to fourfold improvement compared with the endogenous FVIII/VWF complex. The modifications include single-chain reconstruction of the heterodimer of B domain–deleted rFVIII with VWF-D′D3, fusion of the Fc fragment of human immunoglobulin G1, and 2 XTEN polypeptides conjugation.
Endogenous VWF binding to FVIII vs bioengineered BIVV001. (A) Endogenous VWF complexed with FVIII is cleared from plasma with a half-life of 12 to 14 hours. FVIII (blue) binds to VWF via the D′D3 domain (red). VWF multimers are linked by disulfide bonds at the N-terminal D′D3 as well as the C-terminal CK domain (gray). The VWF-A domains (green) bear binding sites for platelet GP Ibα (A1), ADAMTS13 cleavage site (A2), and collagen (A1 and A3). (B) Bioengineered recombinant FVIII (rFVIII)/VWF-D′D3 construct, namely BIVV001, has an extended half-life of 25 to 31 hours, which is a threefold to fourfold improvement compared with the endogenous FVIII/VWF complex. The modifications include single-chain reconstruction of the heterodimer of B domain–deleted rFVIII with VWF-D′D3, fusion of the Fc fragment of human immunoglobulin G1, and 2 XTEN polypeptides conjugation.
FVIII is a cofactor of the intrinsic tenase complex required for active factor IX to cleave the substrate factor X. FVIII binds VWF, and this provides plasma stability and masks FVIII phospholipid binding sites involved in the intrinsic tenase assembly (see figure, panel A). The FVIII binding site on VWF has been defined by many techniques and narrowed down to the VWF-D′D3 domains forming interactions with the FVIII-a3 acidic region and the C1-C2 domains.2-4 Low-resolution electron microscopy (EM) has shown how VWF-D′ extends toward FVIII-C1.2,3 However, none of these reports were of high enough resolution to describe the FVIII/VWF interface in full and characterize the conformational changes that occur upon complex formation.
FVIII is commonly deficient or mutated in patients with hemophilia A. Currently, the mainstay of treatment in the severe cases of hemophilia involves frequent injections (every 2 days) of FVIII/VWF concentrates. Because of the short half-lives of FVIII (∼3 hours) and VWF (∼15 hours), the high frequency required for FVIII replacement therapies places a burden on the patients and the health care system, thereby calling for development of extended half-life products. Fuller et al achieved a remarkable extension of FVIII half-life by exploiting the natural stabilization/masking mechanism from the VWF-D′D3 domains in the setting of a highly sophisticated recombinant heterodimeric protein termed BIVV001. BIVV001 is a tour de force of protein engineering involving multiple modifications that have been previously described, including utilization of a B domain–deleted recombinant FVIII-Fc fusion protein,5 which complexes with a VWF-DʹD3-Fc fusion, giving improved stability of the complex1 (see figure, panel B).
The BIVV001 heterodimer is too large for crystallization studies, but instead Fuller et al1 exploited recent advances in Cryo-EM as a means to determine the 3-dimensional (3D) structure. The Cryo-EM camera used was the revolutionary Gatan K3 direct electron detector, and the 2017 Nobel Prize in Chemistry was awarded for developments in the utility of this technique for resolving 3D structures of complex macromolecules. Thus, it is interesting that a recombinant form of FVIII engineered principally to increase FVIII plasma lifetime for patients with hemophilia also facilitated a new 3D structure determination. The BIVV001 structure now defines in detail the FVIII/VWF interfacial interactions, burying a large surface area of 2480 Å2 across 5 interaction interfaces. The structure brings the molecular basis of hemophilia A and type 2N von Willebrand disease mutations into focus, as a majority of them reside in the FVIII/VWF interface, disrupting the interaction, thereby causing premature degradation of FVIII/VWF. Newly described in the BIVV001 structure is the role of the sulfated residue tyrosine 1680 in the FVIII-a3 acidic peptide. The sulfated tyrosine 1680 side chain extends into the interface between FVIII-C1 and VWF-D′ domains. The tyrosine 1680 sulfate forms a series of interactions, including a salt bridge to arginine 816 in the basic groove of VWF-Dʹ, and hydrogen bonds to serine 2119 and threonine 2120 of FVIII, making full use of the posttranslational modification as a facilitator of this protein interaction.
A second interesting feature of the study compared the new BIVV001 structure with existing crystal structures reported for the isolated human FVIII and human/porcine chimera FVIII. They were also able to compare the BIVV001 VWF-D′D3 structure with the crystal structure of VWF-D′D3 reported recently, which also contained the cysteine residues required for VWF-D3 dimerization mutated to alanine.6 Although they observed good agreement with the FVIII structures and only minor conformational changes, there was a very large conformational change for VWF-D′D3 whereby VWF-D3 rotates nearly 85° relative to Dʹ around a “hinge” point. The large difference in the VWF-D′D3 conformation points to a high level of flexibility in the VWF structure and could be due to the low pH conditions under which the VWF-D′D3 crystal structure was determined. Thus, the conformation of VWF-D′D3 within the BIVV001 structure may more closely represent the plasma pH disulfide–linked dimer of the VWF-D3 structure. Therefore, a missing piece of the puzzle is still the disulfide-linked VWF-D3 dimer structure, which is not described by the BIVV001 structure. A hint at what this may look like comes from the recently described Cryo-EM structures of mucins, which share the same D′D3 disulfide-linked dimer domain architecture as in VWF.7
One other property of VWF-D′D3 that is also not fully understood is the relationship with the VWF-A1 domain. It is well known that VWF has a shear force–activated structure whereby binding sites of platelet glycoprotein (GP) Ibα (A1 domain), ADAMTS13 (A2 domain), and collagen (A1 and A3 domains) become exposed. When VWF is released from activated endothelial cells, it folds into a coiled conformation in which the platelet GP Ibα binding site in the VWF-A1 domain is shielded via an unknown mechanism, involving D′D3.8 ,9 Thus, there may be a linkage between the VWF-D′D3 conformational changes associated with FVIII binding and the VWF-A1 shielding mechanism. Cryo-EM now makes it possible to visualize very large macromolecular complexes, the bigger the better, in principle. This may allow ultimately for the structures of the native dimeric FVIII/VWF-D′D3 complex and VWF-A1A2A3 assembly to be also “caught on camera.”
Conflict-of-interest disclosure: The authors declare no competing conflicts of interest.

