

Key Points
VWF-D′ makes extensive interactions with FVIII; visualization of FVIII-a3 showcases a central role of sulfated Y1680 in binding VWF R816.
The structure of BIVV001 provides insights into the regulation of the FVIII-VWF complex and disease-causing mutations.

Abstract
Interaction of factor VIII (FVIII) with von Willebrand factor (VWF) is mediated by the VWF D′D3 domains and thrombin-mediated release is essential for hemostasis after vascular injury. VWF-D′D3 mutations resulting in loss of FVIII binding are the underlying cause of von Willebrand disease (VWD) type 2N. Furthermore, the FVIII–VWF interaction has significant implications for the development of therapeutics for bleeding disorders, particularly hemophilia A, in which endogenous VWF clearance imposes a half-life ceiling on replacement FVIII therapy. To understand the structural basis of FVIII engagement by VWF, we solved the structure of BIVV001 by cryo-electron microscopy to 2.9 Å resolution. BIVV001 is a bioengineered clinical-stage FVIII molecule for the treatment of hemophilia A. In BIVV001, VWF-D′D3 is covalently linked to an Fc domain of a B domain–deleted recombinant FVIII (rFVIII) Fc fusion protein, resulting in a stabilized rFVIII/VWF-D′D3 complex. Our rFVIII/VWF structure resolves BIVV001 architecture and provides a detailed spatial understanding of previous biochemical and clinical observations related to FVIII–VWF engagement. Notably, the FVIII acidic a3 peptide region (FVIII-a3), established as a critical determinant of FVIII/VWF complex formation, inserts into a basic groove formed at the VWF–D′/rFVIII interface. Our structure shows direct interaction of sulfated Y1680 in FVIII-a3 and VWF-R816 that, when mutated, leads to severe hemophilia A or VWD type 2N, respectively. These results provide insight on this key coagulation complex, explain the structural basis of many hemophilia A and VWD type 2N mutations, and inform studies to further elucidate how VWF dissociates rapidly from FVIII upon activation.
Introduction
Factor VIII (FVIII) is a lynchpin of the coagulation cascade whose spatial and temporal regulation ensures hemostasis under normal conditions and cessation of bleeding upon vascular injury. Alone, FVIII survives in the bloodstream for only 3 hours, whereas complexation with its carrier protein, von Willebrand factor (VWF), extends its half-life to 12 to 14 hours in humans.1-3 Biochemical, animal model, and clinical data have contributed to our current understanding of the FVIII/VWF complex.4-7 VWF-D′D3 binds multiple sites on FVIII, including C1, C2, a3, and potentially a portion of A3 (Figure 1A-B). VWF-D′D3 is sufficient for FVIII binding and Dʹ contributes the majority of the interaction’s affinity.8 Occlusion of the FVIII C domains by VWF-D′D3 prevents interaction of FVIII with phospholipid membranes and is implicated in preventing premature FVIII clearance. Upon initiation of coagulation, thrombin cleaves FVIII at positions R372, R740, and R1689, located in the FVIII acidic regions a1, a2, and a3, respectively (Figure 1A).9-11 Cleavage releases VWF from FVIII, exposing the FVIII C domains for association with anionic phospholipid surfaces and allowing formation of the intrinsic tenase complex.12 The role of sulfated Y1680 in FVIII-a3 has gained the most attention.13,14 FVIII-VWF interaction is mediated by FVIII-a3 region and is severely compromised when Y1680 within FVIII-a3 is mutated or absent. Impaired FVIII-VWF interaction results in the rapid clearance of FVIII and accompanying bleeding diathesis.14-16 Similarly, mutations in VWF-D′ or VWF-D3 that disrupt association with FVIII result in the bleeding disorder, von Willebrand disease (VWD) type 2N.17,18
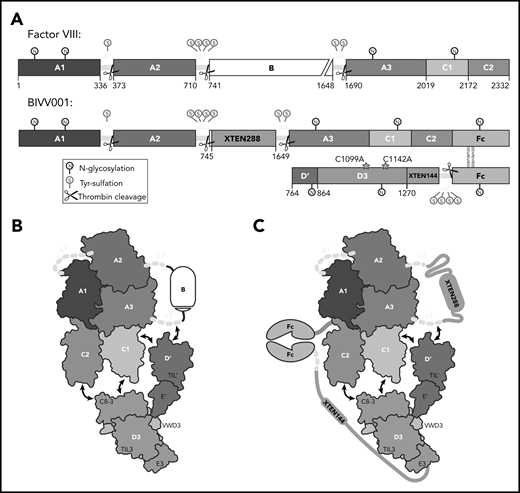
BIVV001, a stabilized FVIII/VWF-DʹD3 complex, was an ideal candidate for single particle, cryo-electron microscopy. (A) N to C terminal linear domain layout comparing WT FVIII (top) to BIVV001 (bottom). Large structured domains of FVIII are in blue, with intervening acidic peptides in yellow, and B domain in white. In BIVV001, XTEN insertions are in gray, Fc in green, and Dʹ and D3 in red and orange, respectively. Positions of C1099A and C1142A mutants to prevent VWF dimerization are shown. Thrombin cleavage sites are depicted by scissors, with posttranslational modification sites indicated by spheres: S, tyrosine sulfation; N, N-linked glycosylation. (B) Illustration of FVIII/VWF-D′D3 complex for WT FVIII and endogenous VWF. (C) Illustration of VWF-D′D3 complex within BIVV001. Arrows depict likely interaction sites based on previously reported biochemical data.
BIVV001, a stabilized FVIII/VWF-DʹD3 complex, was an ideal candidate for single particle, cryo-electron microscopy. (A) N to C terminal linear domain layout comparing WT FVIII (top) to BIVV001 (bottom). Large structured domains of FVIII are in blue, with intervening acidic peptides in yellow, and B domain in white. In BIVV001, XTEN insertions are in gray, Fc in green, and Dʹ and D3 in red and orange, respectively. Positions of C1099A and C1142A mutants to prevent VWF dimerization are shown. Thrombin cleavage sites are depicted by scissors, with posttranslational modification sites indicated by spheres: S, tyrosine sulfation; N, N-linked glycosylation. (B) Illustration of FVIII/VWF-D′D3 complex for WT FVIII and endogenous VWF. (C) Illustration of VWF-D′D3 complex within BIVV001. Arrows depict likely interaction sites based on previously reported biochemical data.
The FVIII-VWF interface has not yet been visualized at high resolution. Previous studies have characterized FVIII/VWF-D′D3 assembly at low resolution by negative stain electron microscopy (EM),4,5 whereas others have probed the interaction using hydrogen deuterium exchange mass spectrometry,5,6 nuclear magnetic resonance,7 and other methods.15,19-21 Multiple structures of FVIII alone have been solved to moderate resolution (≥3.7 Å) over the past decade by X-ray crystallography.22-24 Recently, an atomic resolution X-ray structure of VWF-D′D3 was solved at 2.5 Å resolution,25 and Smith et al concurrently published the highest resolution structure to date (3.2 Å) of an engineered human FVIII variant with improved geometry and sequence register assignments.26 These 2 structures, in combination with the previous low-resolution EM envelope,4 enable construction of FVIII/D′D3 complex docking models. Although physiologically important, the FVIII-a3 has evaded visualization and atomic resolution detail for the FVIII/VWF-D′D3 complex is unknown.
A detailed understanding of the FVIII/VWF complex is highly relevant to development of therapeutics for the treatment of hemophilia A. Extended half-life (EHL) FVIII replacement therapies are recombinant FVIII (rFVIII) variants with modifications (eg, fusion to human immunoglobulin G1 Fc domain, conjugation to polyethylene glycol) that increase FVIII half-life.27 EHL product FVIII half-lives are constrained to an 18- to 19-hour ceiling imposed by VWF-mediated clearance27,28 ; a 1.5- to twofold half-life improvement over the endogenous FVIII/VWF complex. Evolving treatment goals in hemophilia, such as long-term joint protection, and improved patient-reported quality of life require high sustained FVIII levels.29,30 BIVV001, a novel fusion protein consisting of single-chain human B domain–deleted rFVIII, 2 immunoglobulin G1 Fc domains, VWF-D′D3 domains, and 2 XTEN (Amunix Pharmaceuticals, Inc, Mountain View, CA) polypeptide linkers, was designed to overcome the VWF-mediated half-life ceiling (Figure 1A-C). In mice and monkeys, BIVV001 FVIII half-life is 25 to 31 hours and 33 to 34 hours, respectively, which is a three- to fourfold increase over native FVIII in these species.31 Early clinical data suggest a single dose, and 4 once-weekly doses, of BIVV001 are well tolerated and achieve high sustained FVIII activity (near normal at day 3 and 10% at day 7 after dosing) in subjects with severe hemophilia A.32,33
We hypothesized that the engineered stable interaction between FVIII/VWF-D′D3 complex in BIVV001 provided by an intervening Fc fusion domain would enable high-resolution structural determination of this highly sought-after coagulation complex. We used cryo-electron microscopy (cryo-EM) to solve the structure of BIVV001 to 2.9 Å. The structure shows the global architecture by which VWF shields FVIII from phospholipid binding and subsequent degradation in vivo, and details the interaction of FVIII C domains and FVIII-a3 with VWF-D′D3. From the analysis of these interactions, a 2-step binding model for VWF to FVIII is proposed. Our results further detail the probable molecular basis by which many known mutations, including at residues FVIII-Y1680 and VWF-R816, lead to hemophilia A or VWD type 2N. These results provide a structural basis for previous biochemical and clinical observations and raise new questions about how VWF modulates FVIII clearance and how thrombin cleavage at R1689 in FVIII-a3 results in dissociation of activated FVIII from VWF.
Methods
Protein production
BIVV001 was produced as described previously.31 Briefly, a human B domain–deleted rFVIII-Fc-XTEN construct and a VWF (D1-D3 domains/C1099A/C1142A)-XTEN-Fc construct were cloned into separate expression vectors using standard molecular biology techniques (Figure 1A). Expression was performed in HEK293 cells grown in serum-free suspension culture. Cells were cotransfected with the 2 BIVV001 component plasmids and a third plasmid coding expressing PACE/furin for intracellular cleavage of the VWF-D1D2 propeptide. The resulting BIVV001 heterodimer (Figure 1C) was purified by affinity chromatography using VIIISelect resin followed by additional chromatography steps.
Cryo-EM grid preparation
BIVV001 was buffer-exchanged into 25 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid pH 7.3, 150 mM NaCl, and 2.5 mM CaCl2 by gel filtration chromatography using Superose 6 Increase resin (GE Healthcare). Peak fractions containing BIVV001 were pooled and concentrated to 0.75 mg/mL (measured by absorbance at 280 nm). NP-40s detergent was added to samples immediately before grid freezing, to a final concentration of 0.0038% (w/v).
Cryo-EM exposures were collected from 3 distinct grids (supplemental Table 1, available on the Blood Web site) and pooled for processing and analysis. A PELCO easiGlow device was used to plasma clean the grids before sample application, and a Vitrobot Mark IV System (Thermo Fisher Scientific) with the chamber held at 100% humidity and 18°C was used for plunge freezing. A 3.5-µL droplet of BIVV001 sample was dispensed onto the grids, blotted for 5 seconds, and immediately plunge frozen in liquid ethane. Additional details are available in the supplemental Methods.
Cryo-EM data collection and processing
All exposures (supplemental Table 1) were collected using the same Titan Krios transmission electron microscope (Thermo Fisher Scientific) equipped with a BioQuantum energy filter and K3 direct electron detector (Gatan) operating in superresolution mode. Calibrated physical pixel size at the detector was 1.06 Å. One dataset was collected with the specimen stage tilted to 30°. For each movie exposure, patch-based motion correction, binning of superresolution pixels, and frame dose-weighting was performed using RELION-3.0.34 Contrast transfer function estimation was performed on non–dose-weighted sums using CTFFind 4.1.13.35 Micrographs that were outliers in either motion or contrast transfer function parameters were discarded. The cisTEM software package36 was used for particle picking, 2-dimensional (2D) classification, and ab initio 3-dimensional (3D) model generation. Particle positions were picked using a minimum interparticle distance of 144 Å and a template radius of 40 Å. These were subjected to several rounds of 2D classification to remove false particle picks. Following ab initio 3D model generation, 3D refinement in cisTEM was used to refine particle position coordinates.
RELION-3.1 was used for the remainder of data processing, starting from refined particle coordinates. Initial steps used a 3D mask that excluded the poorly resolved VWF-D3. First, 3D autorefinement, per-particle defocus refinement, and beam-tilt refinement were iterated until subjective map quality and reported resolution ceased to improve. Three-dimensional classification using local angular searches was then used to select a subset of particles that yielded a substantially improved map; this subset was further enhanced via per-particle motion correction. To focus additional 3D classification toward improving the FVIII-C1/-C2 and VWF domains, partial signal subtraction was then used to remove the density for most of the FVIII A domains. The 3D classification without angular searches was performed on the subtracted particles, using a mask around the remaining domains (including VWF-D3). Particles from a class showing improved density and connectivity for FVIII-C2 and VWF-D3 were selected, and their nonsubtracted counterparts used in a final round of 3D refinement within a mask including all domains. The final 3D refinement used SIDESPLITTER37 to prevent remaining heterogeneity from degrading global map quality. The final map was sharpened using a fitted B factor and filtered to local resolution for use in atomic model building. The data processing workflow is summarized in supplemental Figure 1, and further details on data processing are given in the supplemental Methods.
Atomic model building
An atomic model for the FVIII A and C1 domains, as well as the majority of the VWF-D′ domain, was built de novo into the EM potential map with the Coot,38 with the available human FVIII (Protein Data Bank [PDB] ID 6mf2)26 and VWF-D′D3 (PDB ID 6n29)25 structures as guides. For these regions, model geometry was further improved using rebuilding tools from the Rosetta Software Suite.39 Following this, FVIII-C2 and VWF-D3 from the PDB IDs mentioned previously were docked into the map using the rigid body fitting functions of UCSF Chimera40 and Coot.38 The geometry of this full model, particularly in regions that could not be built manually, was then refined using the ISOLDE41 plugin to UCSF ChimeraX42 and global real-space model minimization and atomic displacement parameter refinement using the Phenix Software Suite.43 Final model statistics are given in supplemental Table 2. BIVV001 model coordinates have been deposited in the PDB under code 7KWO. Cryo-EM map files have been deposited at the Electron Microscopy Data Bank under code EMD-23057.
Results
Overall structure
Here, we report the 2.9 Å structure of the BIVV001 FVIII/D′D3 complex obtained using single-particle cryo-EM (Figure 2; supplemental Figure 2). The complex exhibited strong preferential orientation for “side” views (Figure 2B; supplemental Figure 3) when vitrified on cryo-EM grids. A combination of mild detergent addition and physical tilt of the sample during data collection was necessary for 3D reconstruction. The particle angular distribution remained highly skewed (supplemental Figure 3); however, a sampling of directional Fourier shell correlations44 show that most angular perspectives exhibit apparent resolution of at least 3.2 Å (supplemental Figure 2). The cryo-EM map provides unambiguous identification of all structured subdomains of FVIII12 and VWF-D′D3, which can be further subdivided into subdomains TIL′ and E′ for VWF-D′ and VWF-D3-C8_3-TIL3-E3 for VWF-D345 (Figure 1B-C). The long axis of D′ lies along the outer edge of FVIII-C1, slightly angled compared with the long/vertical axis of FVIII (Figure 2D-E). The N terminus of D′ reaches up to the base of FVIII-A3 and the D′-D3 junction sits on the same horizontal plane as the bottom of the FVIII C domains, positioned toward the front face of C1. VWF-D3 sits below FVIII, with the thicker end of wedge-shaped D3 positioned between FVIII-C1 and FVIII-C2 at an angle. As expected, the XTEN insertions are not visible in the final map and were not apparent in any 2D class averages. The same is true for the Fc domain; however, in some buffer conditions during preliminary cryo-EM screening, the Fc occasionally adopted a preferred position on the back side of FVIII below the A3 protrusion (supplemental Figure 4).
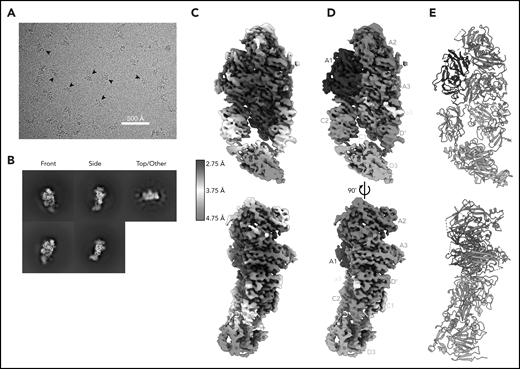
Cryo-EM structure of FVIII/VWF-DʹD3 complex solved to 2.9 Å resolution. (A) Single micrograph of BIVV001 particles at ×81 000 magnification. Black arrows indicate BIVV001 particles. (B) Representative 2-dimensional class averages of BIVV001, demonstrating multiple particle orientations. Side views were readily observed (middle top and middle bottom) and were the preferred orientation; front-on views (left top and left bottom) and top/other views (right) were also found. (C) Local resolution gradient across EM potential map, colored from high (blue, 2.75 Å) to low (red, 4.75 Å). The core of FVIII is the best resolved portion of the structure, with VWF-D3 the least well resolved. (D) EM potential map colored according to protein domain identity. (E) Representation of FVIII/VWF-D′D3 complex structure. Bound metal ions are depicted as spheres.
Cryo-EM structure of FVIII/VWF-DʹD3 complex solved to 2.9 Å resolution. (A) Single micrograph of BIVV001 particles at ×81 000 magnification. Black arrows indicate BIVV001 particles. (B) Representative 2-dimensional class averages of BIVV001, demonstrating multiple particle orientations. Side views were readily observed (middle top and middle bottom) and were the preferred orientation; front-on views (left top and left bottom) and top/other views (right) were also found. (C) Local resolution gradient across EM potential map, colored from high (blue, 2.75 Å) to low (red, 4.75 Å). The core of FVIII is the best resolved portion of the structure, with VWF-D3 the least well resolved. (D) EM potential map colored according to protein domain identity. (E) Representation of FVIII/VWF-D′D3 complex structure. Bound metal ions are depicted as spheres.
The cryo-EM map varies widely in local resolution. Density of sufficient quality and resolution for de novo model building was readily observed for the A1, A2, A3, a3, and C1 domains of FVIII and for most of VWF-D′ (Figure 2C). Density for FVIII-C2 and VWF-D3 required extensive particle 3D classification to resolve (supplemental Figure 1) and was observed at a lower resolution that was not suitable for manual model building or refinement (Figure 2C). Published X-ray structures25,26 were unambiguously docked into these regions based on shape and secondary structure features (supplemental Figure 5). In our model, these domains were not built manually but refined computationally. Upon docking, all subdomains of VWF-D3 were observed in density for BIVV001, except for the most terminal VWF-D3 E3 subdomain (Figure 1B-C).
The FVIII, VWF-D′, and VWF-D3 components of the BIVV001 show no gross morphological changes from previous crystal structures (the only notable difference is the orientation of D′ relative to D3), indicating BIVV001 represents the native FVIII/VWF complex. In particular, our map and model agree with the amino acid register shift(s), N-linked glycosylation sites, and metal binding sites in the improved Smith et al26 human FVIII model, with an overall root-mean-square deviation of 0.74 Å over 1070 aa (Figure 3). Notably, though, our atomic model exhibits considerably better validation and quality metrics (geometry outliers, clashscore, MolProbity46 score) than all other available FVIII structures, most likely resulting from the better resolution of our map (supplemental Table 2). VWF-D′ in our model also superimposes well with the previously published crystal structure with an root-mean-square deviation of 2.2 Å over 98 aa, with slight rotation of E′ relative to TIL′ (Figure 3B).
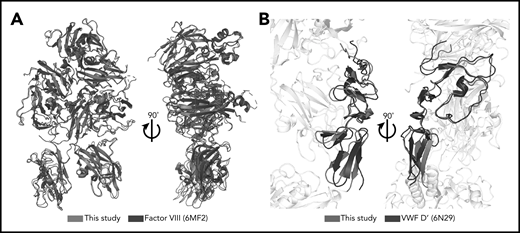
Minimal structural differences exist between complexed FVIII and VWF-D′ compared with free FVIII and VWF-D′. (A) Superposition of complexed FVIII from this study (blue) and free FVIII from PDB: 6MF2 (black). (B) Superposition of complexed VWF-D′ from this study (red) and free VWF-D′ from PDB: 6N29 (black). The FVIII and VWF-D3 portions of the structure are in gray.
Minimal structural differences exist between complexed FVIII and VWF-D′ compared with free FVIII and VWF-D′. (A) Superposition of complexed FVIII from this study (blue) and free FVIII from PDB: 6MF2 (black). (B) Superposition of complexed VWF-D′ from this study (red) and free VWF-D′ from PDB: 6N29 (black). The FVIII and VWF-D3 portions of the structure are in gray.
For much of FVIII-C2 and the entirety of VWF-D3, the local resolution of our map is limited. Map clarity for the C2 domain improved considerably following 3D classification (supplemental Figure 1), which indicates that the domain as a whole may move relative to the rest of FVIII, especially when not interacting with VWF-D3. Similarly, Smith et al observed a novel conformation for the C2 domain (“model B”) in their engineered FVIII crystal structure.26 However, after 3D classification to enhance FVIII-C2, VWF-D3, and surrounding regions, our final map unambiguously places FVIII-C2 in a pose very similar to previous human FVIII crystal structures (as opposed to the Smith et al “model B”) when engaged with VWF-D3. In the final map, local resolution for the C2 domain still degrades rapidly in the distal half of the domain (Figure 2C; supplemental Figure 5), which suggests internal flexibility, including in the loops that likely interact with VWF-D3, is also present. The local resolution around VWF-D3 itself is even worse, indicating that its positioning is even more heterogeneous. Evaluation of the internal structure of this domain is thus limited to gross morphology, which does not appear to deviate from the previous X-ray structure (supplemental Figure 5).
The total buried surface area of VWF-D′D3 with FVIII is 2480 Å2 across 5 interaction interfaces, including the previously unobserved FVIII-a3 (Table 1).
Details of interaction interfaces between FVIII and VWF
| Interface | Surface area (Å2) | No. of hydrogen bonds/salt bridges | |
|---|---|---|---|
| Global interactions | |||
| FVIII | VWF-D′D3 | 2486 | 22/12 |
| FVIII | VWF-D′ | 1817 | 19/9 |
| FVIII | VWF-D3 | 669 | 3/3 |
| Detailed interactions | |||
| FVIII C1 | VWF-D′ | 943 | 7/2 |
| FVIII-a3 peptide | VWF-D′ | 480 | 11/7 |
| C2 | VWF-D3 | 472 | 2/2 |
| A3 | VWF-D′ | 394 | 1/0 |
| C1 | VWF-D3 | 197 | 1/1 |
| FVIII-a3 peptide | FVIII-A3* | 84 | 0/0 |
| FVIII-a3 peptide | FIII-C1* | 81 | 3/0 |
| Interface | Surface area (Å2) | No. of hydrogen bonds/salt bridges | |
|---|---|---|---|
| Global interactions | |||
| FVIII | VWF-D′D3 | 2486 | 22/12 |
| FVIII | VWF-D′ | 1817 | 19/9 |
| FVIII | VWF-D3 | 669 | 3/3 |
| Detailed interactions | |||
| FVIII C1 | VWF-D′ | 943 | 7/2 |
| FVIII-a3 peptide | VWF-D′ | 480 | 11/7 |
| C2 | VWF-D3 | 472 | 2/2 |
| A3 | VWF-D′ | 394 | 1/0 |
| C1 | VWF-D3 | 197 | 1/1 |
| FVIII-a3 peptide | FVIII-A3* | 84 | 0/0 |
| FVIII-a3 peptide | FIII-C1* | 81 | 3/0 |
FVIII-FVIII interaction.
FVIII-a3 peptide interactions
Our high-resolution cryo-EM map provides detail on previously unobserved structural elements of FVIII-VWF engagement, notably the FVIII-a3 peptide (Figure 4). We observe well-resolved sidechain density for a 7-residue stretch of the FVIII-a3 peptide (E1675-E1681) and clear density for sulfation of Y1680 (supplemental Figure 6), which buries 480 Å2 against VWF-D′. The FVIII-a3 peptide and sulfation of FVIII-Y1680 are critical for the FVIII-VWF interaction in vivo.13,47 Our model shows sulfated FVIII-a3 Y1680 interacting directly with the sidechain of VWF-D′ R816. Mutation of either residue causes disease, with mutations in FVIII-Y1680 resulting in mild or moderate hemophilia A,13,47 and mutations in VWF-D′-R816 resulting in the most severe form of VWD type 2N.18,48,49 FVIII residues H1867 in A3 and S2119 in C1, mutation of which causes hemophilia A,50 are part of the binding pocket for the sulfated Y1680 sidechain (Figure 4B). FVIII-a3 also binds along a groove in VWF-TIL′ as a third β-strand to the VWF-TIL′ β sheet (supplemental Figure 7), consistent with recent nuclear magnetic resonance data.7 The groove is highly basic and complements the acidic nature of FVIII-a3 (Figure 4A), as predicted by Dagil et al.7 Specifically, FVIII acidic a3 residues D1676 and D1678 form interdigitating salt bridges with VWF-D′ R820 and R826 on the same face of the β sheet, along with another charged interaction between FVIII-a3 D1681 and D′ R768 on the opposite side. The remaining 11 residues that span the thrombin activation site R1689 and connect this portion of FVIII-a3 to FVIII-A3 are not visible.
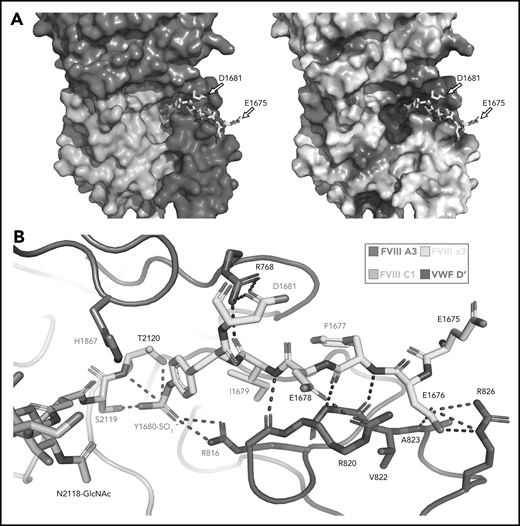
Sulfated-Y1680 in the FVIII-a3 acidic peptide and R816 in the basic groove of VWF-D′ directly interact. (A) FVIII-a3, shown in yellow with acidic residues labeled, sits in a basic groove of VWF-D′ formed by a network of arginine residues. VWF-D′ is rendered in surface view colored by domain identity (left) or electrostatic potential (right) (acidic: red, basic: blue, neutral: white) using the APBS plugin in PyMol. Scale is –5 to +5 kT/e. (B) Details of FVIII-a3/VWF-D′ interface. FVIII-a3 peptide in yellow, VWF-D′ in red, FVIII-C1 in cyan, and FVIII-A3 in blue. Direct polar interactions are indicated by dotted lines, with interacting residues labeled. Residues with known mutations that cause bleeding disorders are labeled in red.
Sulfated-Y1680 in the FVIII-a3 acidic peptide and R816 in the basic groove of VWF-D′ directly interact. (A) FVIII-a3, shown in yellow with acidic residues labeled, sits in a basic groove of VWF-D′ formed by a network of arginine residues. VWF-D′ is rendered in surface view colored by domain identity (left) or electrostatic potential (right) (acidic: red, basic: blue, neutral: white) using the APBS plugin in PyMol. Scale is –5 to +5 kT/e. (B) Details of FVIII-a3/VWF-D′ interface. FVIII-a3 peptide in yellow, VWF-D′ in red, FVIII-C1 in cyan, and FVIII-A3 in blue. Direct polar interactions are indicated by dotted lines, with interacting residues labeled. Residues with known mutations that cause bleeding disorders are labeled in red.
VWF-TIL′ interactions with FVIII-C1 and -A3 domains
In addition to interactions with the FVIII-a3 peptide, VWF-D′-TIL′ makes extensive interactions with FVIII-C1 and FVIII-A3, consistent with previous studies on FVIII/VWF,4-6,51 burying 987 Å2 and 386 Å2 of surface area, respectively. The VWF-TIL′/FVIII-C1 and VWF-TIL′/FVIII-A3 interfaces appear to be driven largely by Van der Waals interactions and shape complementarity (Figure 5A-B). The N terminus of D′ inserts into a pocket of the FVIII-A3 domain, as predicted by Smith et al, and supports the role of S764 at the N terminus of mature VWF in contributing to the interaction with FVIII.21,26 Below the VWF-D′-TIL′ subdomain, which seems to anchor VWF to FVIII, VWF-D′-E′ makes only minimal contacts with FVIII and appears to act as a connector between VWF-D′-TIL′ and VWF-D3, consistent with previous work4 and supported by the lack of VWD type 2N mutations in this region. In contrast to previous hypotheses, the TIL′ does not make any contacts with FVIII-C2.4
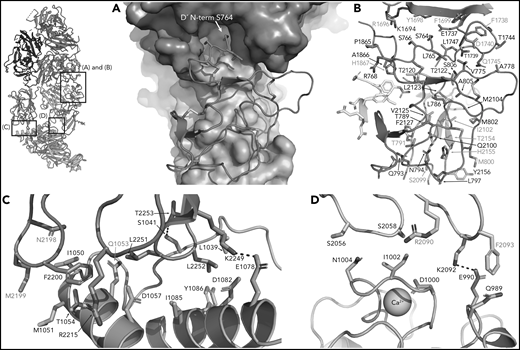
FVIII/VWF-D′D3 form an extended interface of van der Waals interactions. (A) VWF-TIL′ (red) exhibits shape complementarity to FVIII, C1 (cyan), and A3 (violet) domains shown in surface rendering. Insertion of the VWF-TIL′ N terminal residue in a shallow pocket in the A3 domain can be seen. (B) VWF-TIL′/FVIII interacting residues, outside of the a3 peptide, shown as sticks, colored as in panel A. Residues labeled in red have known disease-causing mutations. (C) Docking solution of VWF-D3 indicates C8-3 subdomain (orange) is in close contact with the FVIII C2 domain (teal), with hydrophobic spike residues in FVIII-C2 shown as sticks. (D) The calcium-binding site of VWF-D3 VWD3 subdomain (orange) is in close proximity to the FVIII C1 domain (cyan).
FVIII/VWF-D′D3 form an extended interface of van der Waals interactions. (A) VWF-TIL′ (red) exhibits shape complementarity to FVIII, C1 (cyan), and A3 (violet) domains shown in surface rendering. Insertion of the VWF-TIL′ N terminal residue in a shallow pocket in the A3 domain can be seen. (B) VWF-TIL′/FVIII interacting residues, outside of the a3 peptide, shown as sticks, colored as in panel A. Residues labeled in red have known disease-causing mutations. (C) Docking solution of VWF-D3 indicates C8-3 subdomain (orange) is in close contact with the FVIII C2 domain (teal), with hydrophobic spike residues in FVIII-C2 shown as sticks. (D) The calcium-binding site of VWF-D3 VWD3 subdomain (orange) is in close proximity to the FVIII C1 domain (cyan).
VWF-D3 interactions with FVIII C domains
VWF-D3 is angled below the FVIII-C1 and -C2 domains, burying 169 Å2 and 455 Å2 of surface area, respectively. The position of VWF-D3 relative to FVIII is in good agreement with previous negative stain EM envelopes.4,5 The relatively sparse interactions observed in our structure between VWF-D3 and the FVIII C domains is consistent with lack of density observed in this region in Yee et al’s 3D reconstruction of FVIII-[D′D3]2 and reflects greater structural heterogeneity in this interface. The most notable feature of VWF-D3 is that, to reach its position in our model from its conformation in the uncomplexed D′D3 crystal structure, D3 must rotate nearly 85° relative to D′ around the “hinge” point connecting these domains25 (Figure 6; supplemental Movie). When complexed with FVIII, VWF-D3-VWD3 subdomain contacts the base of the FVIII-C1 and VWF-D3-C8-3 subdomain binds to the bottom of the FVIII-C2 domain (Figure 2D-E). Specific side chains likely involved in this interaction are shown in Figure 5C-D. Our docking solution places the previously identified conserved Ca2+-stabilized loop in VWD325 as part of the interface with FVIII-C1. By occupying this position, VWF-D3 would sterically block the FVIII C domains from interacting with membrane phospholipids. Finally, the position of VWF-D3 places the dimerization cysteines 1099 and 1142, mutated to alanine here, in a position that is amenable to dimer formation. As described in Dong et al, local conformational changes in VWF-D3 would still be required to expose these residues to promote dimer formation.25
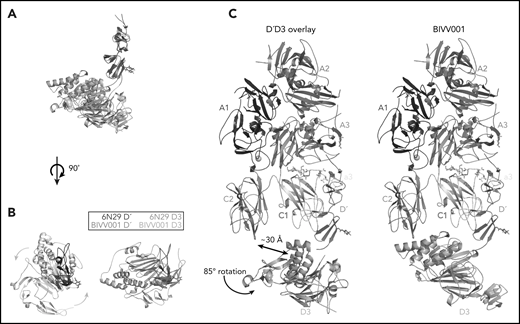
The relative orientation between VWF-D′ and D3 in BIVV001 is rotated compared with the previously reported X-ray structure of VWF-D′D3 alone. (A) Superposition of the VWF-D′D3 X-ray structure (PDB: 6N29), via D′, onto the D′D3 portion of the BIVV001 structure. (B) The individual structures from panel A viewed from the top, placing the long axis of D′ pointed out of the page. The 85° relative rotation of VWF-D3 is indicated by the gradient arrows. (C) The same superposition and transition shown in in the context of the full BIVV001 structure. VWF-D3 rotates 85°, moving the C8-3 α-helices 30 Å to contact FVIII C2.
The relative orientation between VWF-D′ and D3 in BIVV001 is rotated compared with the previously reported X-ray structure of VWF-D′D3 alone. (A) Superposition of the VWF-D′D3 X-ray structure (PDB: 6N29), via D′, onto the D′D3 portion of the BIVV001 structure. (B) The individual structures from panel A viewed from the top, placing the long axis of D′ pointed out of the page. The 85° relative rotation of VWF-D3 is indicated by the gradient arrows. (C) The same superposition and transition shown in in the context of the full BIVV001 structure. VWF-D3 rotates 85°, moving the C8-3 α-helices 30 Å to contact FVIII C2.
Discussion
The cryo-EM structure described here represents the first detailed visualization of the FVIII/VWF-DʹD3 complex. This structure sheds light on the underlying biology of this tightly regulated coagulation complex as well as BIVV001, a VWF-independent FVIII replacement therapy that provides high, sustained FVIII activity.31 This structure is consistent with and provides a structural basis for previous biochemical and clinical reports, bringing decades of data into focus in a single snapshot. Comparing BIVV001 to individual FVIII and VWF-D′D3 structures confirms that BIVV001 is correctly folded and maintains native glycosylation patterns, disulfide bonding networks, and native metal binding sites. The XTEN and Fc of BIVV001 are not visible, demonstrating the fusions are flexible and do not affect native complex integrity. The extensive interfaces of this interaction explain how the VWF-D′D3 domains of BIVV001 shield it from binding free D′D3 in vitro and full-length VWF in vivo, thereby decoupling BIVV001 from endogenous VWF and overcoming the VWF-mediated FVIII half-life ceiling.31
One of the most anticipated findings7,14-16 is the structural detail surrounding FVIII-a3 and its interaction with the VWF-TIL′. Our structure confirms the basic groove on VWF-TIL′ as the primary binding site for FVIII-a3 and underscores the biological relevance of sulfation of FVIII-Y1680 in mediating this interaction.7,13,14,48 Similarly, the structure also provides a molecular rationale for why the VWF R816W mutation leads to the most severe form of VWD type 2N. The location of these and many other critical residues, which when mutated result in the phenotypically similar bleeding disorders of hemophilia A (FVIII) or VWD type 2N (VWF), are shown in Figure 7. The structure provides an explanation for the subset of hemophilia A mutations and majority of VWD type 2N mutations25 that fall in the interface with VWF-D′D3; these mutations likely cause disease, at least in part, owing to the disruption of the FVIII-VWF interaction and subsequent rapid FVIII degradation (supplemental Tables 3 and 4).
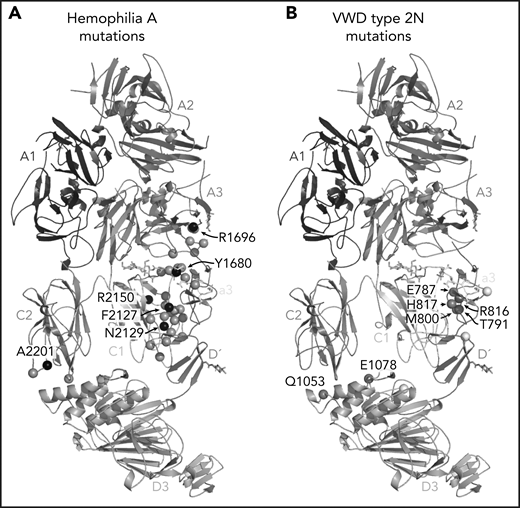
Hemophilia A mutations in FVIII and VWD type 2N mutations in VWF-D′D3 decorate the protein-protein interface. (A) Hemophilia A missense mutations in the interface with VWF are shown on FVIII as gray spheres, with prevalent hemophilia A mutations labeled and depicted as black spheres (defined here as >10 cases reported in the European Association for Haemophilia and Allied Disorders F8 variant database.50 (B) VWD type 2N mutations, as annotated in Dong et al,25 are shown on VWF-D′D3. Mutations in gray are in the interface with FVIII. Mutations in white are outside the FVIII interface, as annotated by PDB PISA. The calcium ion in VWF-D3 is not shown.
Hemophilia A mutations in FVIII and VWD type 2N mutations in VWF-D′D3 decorate the protein-protein interface. (A) Hemophilia A missense mutations in the interface with VWF are shown on FVIII as gray spheres, with prevalent hemophilia A mutations labeled and depicted as black spheres (defined here as >10 cases reported in the European Association for Haemophilia and Allied Disorders F8 variant database.50 (B) VWD type 2N mutations, as annotated in Dong et al,25 are shown on VWF-D′D3. Mutations in gray are in the interface with FVIII. Mutations in white are outside the FVIII interface, as annotated by PDB PISA. The calcium ion in VWF-D3 is not shown.
The extensive interactions of VWF-TIL′ with FVIII confirm VWF-D′ is the high-affinity site for FVIII binding within DʹD3.8 Although the D′ of VWF accounts for >1800 Å2 of buried surface area, the ∼twofold larger D3 accounts for only ∼600 Å2. Similarly, the VWF-D′ region of our cryo-EM map matches the quality of the FVIII region indicating a rigid interaction, whereas the local resolution of our map in and around VWF-D3 is significantly lower, indicating D3 may experience continuous motion around its consensus position. This is consistent with previously published EM envelopes and negative stain data, which show rotation of D3 relative to FVIII/D′ and FVIII/D′D3 complex dimers that exhibit an array of angles along the long axes of FVIII.4,5 In our structure of the FVIII/VWF-D′D3 complex, VWF-D3 rotates 85° around the vertical axis of the VWF-D′ domain (Figure 6A-B), relative to the apo VWF-D′D3 crystal structure, to dock beneath the FVIII C domains (Figure 6C). This led us to propose a 2-step binding model in which VWF-TIL′ first initiates high-affinity binding to FVIII-A3/-a3/-C1, followed by rotation of VWF-D3 until it binds FVIII-C1/-C2 to stabilize the conformation reported here. Although there may be an entropic penalty associated with restricting rotation of D3 relative to D′ upon binding FVIII, we hypothesize that this is could be offset by favorable energetics associated with the formation of extensive hydrogen bonds and salt bridges between VWF-D′ and FVIII-a3 (Table 1). In contrast, when thrombin cleaves FVIII-a3, it likely remains bound to D′D3, but the remainder of the interactions between VWF and FVIII outside of FVIII-a3 are not strong enough to offset the entropic penalty of restricting VWF-D′D3 mobility, leading to rapid dissociation from FVIII.
VWF essential functions include regulating FVIII catabolism, preventing premature interaction of FVIII with the phospholipid membrane and protecting FVIII from proteolytic degradation.28 The relative contributions of the FVIII-C1 and FVIII-C2 domains for binding to phospholipids have been extensively investigated. Early reports described the involvement of 4 hydrophobic residues (M2199, F2200, L2251, L2252) critical for membrane interaction in FVIII-C2.52 Later work showed that FVIII-C1 contributes to platelet binding, suggesting a synergistic effect of the C1 and C2 domains in mediating activated FVIII–membrane interactions. Residues K2092 and F209353,54 have been identified as key mediators of the C1–membrane interaction, though others have been reported as well.55,56 Although local resolution at the VWF-D3/FVIII-C1/-C2 interface is poor, contacts between VWF-D3 and the 2 hydrophobic spikes of FVIII-C2, including M2199, F2200, L2251, and L2252, are visible (Figure 5C), as are contacts between K2092 and F2093 from FVIII-C1. Although the low resolution of the FVIII/VWF-D3 interface precludes detailed analysis, our docked model suggests the conserved Ca2+ loop in VWF-D3 may be in close proximity to S2056-S2058 in an adjacent loop in FVIII-C1, which has been implicated in phospholipid binding56 (Figure 5D). Together, VWF-D′D3 is positioned in a way to prevent FVIII interaction with phospholipids.
Details of the interactions mediating FVIII uptake by clearance receptors and antigen-presenting cells are complex and less well defined. The role of low-density lipoprotein receptor-related protein 1 (LRP1) on liver and vascular endothelium in mediating FVIII clearance has been well-described.28,57-59 Although the involvement of the FVIII C1 domain, specifically residues K2092/F2093, has been shown,58,60 the exact mechanism of LRP1 binding is unclear; other FVIII residues likely play as yet-unknown roles in recognition by LRP1 or other cell surface receptors. Similar regions on FVIII C1 domain have been implicated in mediating uptake by antigen-presenting cells,61,62 although other receptors are also involved on these cells including CD206 and Siglec-5.63,64 VWF has been shown to prevent both the clearance of FVIII by blocking interaction with LRP1 as well as preventing uptake of FVIII by dendritic cells.58,65 As discussed, residues K2092/F2093 are occluded by VWF-D3 in our model, providing a structural basis for EHL of FVIII while in complex with VWF.
Recently, Batsuli et al showed that the pathogenic effect of anti-C1 antibodies directed against noncoagulant epitopes of FVIII results from disrupting interaction with VWF and subsequent rapid FVIII degradation.66 Our structure supports the hypothesis that this class of inhibitors act via competition with VWF as these group A antibodies67 bind an epitope at the FVIII-C1/VWF-D′ interface distinct from the phospholipid-binding region of FVIII-C1.
Previous studies described a cofactor function for VWF in which VWF enhances the rate of thrombin cleavage at R168968 that, in turn, induces the rapid dissociation of FVIII from VWF. Our structure does not clearly elucidate the mechanisms of these processes; FVIII R1689 is located in a flexible 11-residue loop between FVIII-a3 and FVIII-A3, which is unresolved in our model. Interestingly, R1689 is adjacent to the upper edge of the pocket formed between FVIII-A3 and FVIII-C1 into which the N terminus of VWF-D′ inserts (Figure 5A-B). It is possible that cleavage at FVIII R1689 results in a conformational change in the FVIII-A3/-C1 pocket that prevents the VWF-D′ N terminus from binding, leading to release of VWF. And, as discussed previously, rotation of VWF-D′ relative to VWF-D3 may also contribute to this process. Considering additional reports of distal anti-C2 antibodies affecting cleavage at R1689 and dissociation from VWF,69,70 there is still much we do not understand about the mechanistic details of thrombin activation of FVIII and subsequent release from VWF. Additional work such as a thrombin-activated FVIII structure may be required for a more complete understanding of these processes.
The interaction of FVIII with D′D3 is multifaceted, comprised of multiple distinct interfaces, which collectively result in a high-affinity, tightly regulated complex. Cryo-EM will be an indispensable tool in the study with structural details of other large and complex coagulation assemblies. With the best resolved FVIII structure published to date and atomic level detail of its interaction with VWF now available, the role of disease-causing mutations, notably mutations at this critical interface, can be rationalized and further investigated.
For original data, please e-mail the corresponding author at joseph.batchelor@sanofi.com.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Kangkang Song, Kyounghwan Lee, and Chen Xu at the UMass Medical School Cryo-EM facility for help with EM data collection, and Yifan Cheng and Gabe Lander for technical advice to overcome preferred orientation issues. Editorial assistance was provided by Francis John Golder from JK Associates, Inc, a member of Fishawack Group of Companies.
This study was funded by Sanofi. Sanofi and Sobi reviewed the manuscript.
Authorship
Contribution: J.R.F., K.E.K., N.C.L., and J.D.B. conceived the study; J.R.F. designed and performed the experiments; J.R.F., K.E.K., N.C.L., R.T.P., and J.D.B. analyzed the results; J.R.F., K.E.K., N.C.L., and J.D.B. wrote the manuscript; and all authors reviewed and approved the final version of the manuscript.
Conflict-of-interest disclosure: K.E.K., N.C.L., R.T.P., and J.D.B. are employees and shareholders of Sanofi. J.R.F. is a former employee of Sanofi.
Correspondence: Joseph D. Batchelor, Sanofi, 225 2nd Ave, Waltham, MA 02451; e-mail: joseph.batchelor@sanofi.com.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal