

Key Points
The fVIII/anti–C1 domain antibody complex highlights the inhibitor epitopes that elicit a pathogenic immune response.
The complex results in a novel, large-scale conformational change of the C2 domain.

Abstract
Antibody inhibitor development in hemophilia A represents the most significant complication resulting from factor VIII (fVIII) replacement therapy. Recent studies have demonstrated that epitopes present in the C1 domain contribute to a pathogenic inhibitor response. In this study, we report the structure of a group A anti–C1 domain inhibitor, termed 2A9, in complex with a B domain–deleted, bioengineered fVIII construct (ET3i). The 2A9 epitope forms direct contacts to the C1 domain at 3 different surface loops consisting of Lys2065-Trp2070, Arg2150-Tyr2156, and Lys2110-Trp2112. Additional contacts are observed between 2A9 and the A3 domain, including the Phe1743-Tyr1748 loop and the N-linked glycosylation at Asn1810. Most of the C1 domain loops in the 2A9 epitope also represent a putative interface between fVIII and von Willebrand factor. Lastly, the C2 domain in the ET3i:2A9 complex adopts a large, novel conformational change, translocating outward from the structure of fVIII by 20 Å. This study reports the first structure of an anti–C1 domain antibody inhibitor and the first fVIII:inhibitor complex with a therapeutically active fVIII construct. Further structural understanding of fVIII immunogenicity may result in the development of more effective and safe fVIII replacement therapies.
Introduction
Hemophilia A is an X-linked bleeding disorder that affects 1 in 5000 males worldwide due to a deficiency in blood coagulation factor VIII (fVIII), an essential protein cofactor for the serine protease factor IXa (fIXa).1,2 Treatment of hemophilia A consists of therapeutic infusions of recombinant or plasma-derived fVIII; however, 30% of severe hemophilia A patients develop a pathogenic immune response that elicits neutralizing alloantibodies (inhibitors), rendering treatment ineffective.3-6 Inhibitor antibodies that recognize the C1 domain of fVIII have been shown to inhibit the ability of fVIII to bind activated platelet surfaces (PSs), form a stable complex with von Willebrand factor (VWF), and undergo endocytosis by antigen-presenting cells.7-9 Hydrogen/deuterium exchange (HDX) epitope mapping defines 2 discrete groups of anti-C1 inhibitors.7 Group A inhibitors possess high binding affinities for fVIII, although they display weak inhibition of fVIII-procoagulant activity, poorly inhibit fVIII binding to PS, and heterogeneously block fVIII binding to VWF.7 By contrast, group B antibodies more strongly inhibit fVIII-dependent procoagulant activity, disrupt fVIII binding to PS and VWF, and prolong thrombin generation in plasma.7-9 Both group A and B inhibitors have also been shown to block fVIII uptake by dendritic cells.7
The monoclonal antibody (mAb) 2A9 is a group A anti–C1 domain inhibitor isolated from a murine hemophilia A model following infusion of human fVIII.7 The 2A9 mAb has been shown to bind with high affinity to human fVIII and modestly compete for both PS and VWF binding, but type II inhibition occurs in which incomplete inhibition is observed at saturating concentrations of antibody. Competition with VWF binding results in increasing the fVIII clearance rate, defining a novel mechanism of antibody inhibitor pathogenicity.7 Mutational analysis of the isolated C1 domain revealed a unique epitope recognized by 2A9 that is distinct from other group A inhibitors.9 To further understand the structural nature of the 2A9 epitope, we determined the structure of the 2A9 antigen-binding fragment (Fab) bound to a bioengineered B domain–deleted (BDD) fVIII therapeutic (ET3i) that harbors a 100% human sequence for the C1 and C2 domains.
Study design
Production of ET3i factor VIII
The BDD human/porcine chimeric fVIII construct ET3i was expressed, purified, and structurally characterized as previously described.10 Recombinant protein production for crystallographic studies was performed in BHK-derived cells as previously described.11 Purified ET3i was subsequently stored in a 50 mM HEPES, pH 7.4, 5 mM CaCl2 and 350 mM NaCl solution at 0.8 mg/mL at −80°C.
Production of 2A9 monoclonal antibody
The 2A9 fVIII C1 domain-specific antibody inhibitor was produced from a hybridoma derived from immunization of hemophilia A mice with intravenous, adjuvant-free injections of human fVIII, as previously described.7 Frozen hybridoma cells were initially thawed at 37°C and centrifuged at 6000 RPM for 2 minutes. The cell pellet was resuspended in 2 mL of Clonacell HY Medium E (Stem Cell Technologies), introduced to T-75 culture flasks and grown at 37°C for 5-7 days at a total volume of 50 mL. At 90% confluence, the cell culture growth media was decanted, and the remaining adherent cells were rinsed 3 times with 3 mL of phosphate buffered saline (PBS). Antibody expression was propagated by the addition of 50 mL of CD Hybridoma Medium (Invitrogen) in the presence of GlutaMax-1, penicillin/streptomycin, and cholesterol. Following 1-week expression, the culture media was decanted, centrifuged at 6000 RPM for 10 minutes, and the antibodies were subsequently purified from the supernatant.
The 2A9 antibody in hybridoma media was diluted 5-fold in MES buffer (adjusted to pH 6) and purified with an SP Sepharose Fast Flow column pre-equilibrated in binding buffer (10 mM MES [pH 6], 200 mM NaCl). The column was subsequently washed with binding buffer until the A280 returned to background levels. Bound 2A9 antibody was eluted with elution buffer (20 mM HEPES [pH 7.4], 150 mM NaCl). A secondary purification step was performed with a protein A+ column. The 2A9 antibody fraction was incubated with protein A+ resin at room temperature for 10 minutes with mixing. The resin was then washed 3 times with SP Sepharose elution buffer, and the 2A9 antibody was eluted with low pH buffer (0.1 M sodium acetate, pH 3), which was immediately neutralized with 1 M Tris-HCl, pH 7.5. The purified, concentrated fractions of the 2A9 antibody were stored at −80°C
Preparation of 2A9 Fab fragments
Purified 2A9 IgG fractions were dialyzed in papain cleavage buffer (10 mM EDTA, 20 mM sodium phosphate, pH 7). Agarose-linked papain (Thermo Scientific) was equilibrated in digestion buffer (papain cleavage buffer, 30 mM cysteine, pH readjusted to 7) and combined with dialyzed 2A9 IgG mixed (1:1) with digestion buffer. The cleavage reaction was incubated at 37°C overnight with rocking. The resultant cleavage reaction was initially clarified by centrifugation with Costar spin filters, and the agarose-linked papain was washed twice with 100 µL of neutralization buffer (50 mM Tris HCl, pH 7.4) and combined with the clarified cleavage fraction. To isolate the purified Fab fractions, the cleavage fraction was flowed over a protein A+ column to separate the Fab and Fc fractions. The eluted, purified 2A9 Fab fractions were stored at −80°C.
Crystallization and structure determination
The ET3i/2A9 Fab complex was initially formed in a (1:1.2) stoichiometric ratio in 50 mM Tris HCl, 200 mM NaCl, 2.5 mM CaCl2, pH 7.4. Initial crystal conditions were determined by high-throughput microbatch crystallization using the Hauptman-Woodward High-Throughput Crystallization Screening Center (Buffalo, NY). Diffraction quality crystals were subsequently grown by hanging drop vapor diffusion in a 1:1 (v/v) ratio of the ET3i/2A9 Fab protein complex with a crystallization solution containing 17.5% (w/v) PEG 1500, 50 mM HEPES (pH 7.7). Crystals were cryoprotected in the same crystallization solution with the addition of 30% (w/v) PEG 400. X-ray diffraction data were collected on the Advanced Light Source (ALS) Berkeley Center for Structural Biology (BCSB) beamline 5.0.1 (Berkeley, CA). X-ray diffraction data collection and processing were performed with Adxv, XDS and CCP4.12 Phasing of the ET3i/2A9 Fab crystals were determined with PHASER-MR by using a fragment-based molecular replacement approach with the previously determined 3.2 Å structure of ET3i (PDBID: 6MF0) and the 2.5 Å structure of the Fab fragment from the IgG2alpha 3E6 antibody (PDBID: 4KI5).13-15 Model building and refinement were performed with Coot and PHENIX, respectively.16 All figure representations were generated with the PyMOL Molecular Graphics System, Version 2.0 (Schrödinger, LLC).
Results and discussion
Structure of fVIII in complex with the anti–C1 domain 2A9 antibody
The X-ray crystal structure of the ET3i BDD fVIII construct in complex with the 2A9 Fab was determined to 3.9-Å resolution and contains each domain of fVIII (A1-A3, C1, C2) as well as the constant and variable domains of the heavy and light chains of 2A9 (Figure 1A; supplemental Table 1, available on the Blood Web site). The overall interface is largely hydrophobic with several aromatic residues and possesses 2429 Å2 of buried surface area. The majority of the binding epitope is within the C1 domain as predicted, and there is a minor binding interface with the A3 domain. The variable domains straddle the Lys2065-Trp2070 surface loop of the fVIII C1 domain (Figure 1B), serving as the centralized region of the epitope for 2A9 and agrees with HDX protection patterns for other group A anti-C1 inhibitors.7 Here, Phe2068 is completely buried at the fVIII/2A9 interface and makes extensive hydrophobic contacts with Phe48, Phe49, Leu45, and Tyr33 of the variable domain of the light chain, forming a “phenylalanine sandwich.” This is consistent with mutational analyses showing that a Phe2068His mutant abrogates 2A9 binding.9 The Arg2150-Tyr2156 loop forms additional contacts between the C1 domain and the variable domains of 2A9 (Figure 1C), with Arg2150 and His2152 making direct contacts with the heavy chain and Thr2154-Tyr2156 making direct contacts with the light chain. These contacts are also substantiated by HDX protection patterns in the presence of 2A9 (supplemental Figure 1).7 Additional C1 domain interactions occur between Lys2110 and Trp2112 with the heavy chain of 2A9. Notably, the intermolecular contacts between the C1 domain and 2A9 in our structure are also involved in the fVIII/VWF D′D3 complex as shown by HDX.17 Minor interactions also occur between Ile2102 and Thr2122 and the heavy and light variable regions, respectively, which are also consistent with minor HDX protection patterns for the VWF D′D3 region. The extent of the overlap between the 2A9 epitope and the VWF-binding interface supports the hypothesis that the mechanism of pathology for group A antibody inhibitors is to increase the clearance of fVIII by disrupting the VWF interaction, dramatically decreasing the plasma half-life of fVIII.7,8 With a Keq and koff of 0.1 nM and 2.2 × 10−4 s−1, respectively, the 2A9 antibody possesses comparable binding characteristics to VWF.17-20 The structure of the 2A9 complex also indicates that the A3 domain contributes to the overall epitope, forming multiple contacts between the Phe1743-Tyr1748 loop of the A3 domain and the 2A9 light chain (Figure 1D). Interestingly, the second N-acetylglucosamine residue at Asn1810 makes electrostatic interactions with Glu1 and Lys95 of the 2A9 light and heavy chains, respectively. The overall structure of the 2A9 antibody bound to fVIII indicates that glycosylation sites potentially contribute to inhibitor development and illustrates that the epitope definitions are more complex than fVIII domain specificity. Moreover, simultaneous recognition of the C1 and A3 domains further supports the physiological relevance of the relative positioning of the C1 and A3 domains in determined fVIII structures.14,21,22
![The structure of the ET3i fVIII/anti–C1 domain 2A9 Fab fragment. (A) Ribbon diagram representation of ET3i in complex with the 2A9 anti–C1 domain Fab antibody fragment (Protein Data Bank identifier [PDBID#] 7K66) (pink, porcine A1 domain; dark magenta, porcine A3 domain; dark teal, human A2, C1, and C2 domains; orange, 2A9 Fab light chain [LC]; green, 2A9 Fab heavy chain [HC]). (B) Direct molecular contacts between the 2A9 antibody and the C1 domain 2065-2070 loop. (C) Direct molecular contacts between the 2A9 antibody and the C1 domain 2050-2056 loop. (D) Surface contacts between the 2A9 antibody and the A3 domain 1743-1748 loop and the second N-acetylglucosamine (NAG) residue N-linked to Asn1810.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/137/21/10.1182_blood.2020008940/1/m_bloodbld2020008940f1.png?Expires=1769283024&Signature=dQmPLO6Xwyc6gsP2nIFlJKdfeS-RczQaaYRQJqaNAo7TALMgz-Ipj8UNkxYxWCYJLMjBWqQqTBgZIzUr39zqgkrEaThCO~xDK6aIMHjyywrsAvI0Yj-xvt4CwUxF1WSG-BGUXp4YECYp9uVQaIw5K5UczByqLgxbS6odhqTXAnQ6Vm4ZxsgjZArvp26xL7ivP4ApxBqPz8I54Y2xa3QyALtDjS8w6WuWLHkv96PL229PWXBV3M7XyZAjn7p3kjKEmMRdtuBsVvjP~l9AvgYXUksF6IWEHTgKOQuvayvhBqmHtrFqb0Nm-VHPIEYCQE~nNVku42qjIvU75dWD2D3WVQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
The structure of the ET3i fVIII/anti–C1 domain 2A9 Fab fragment. (A) Ribbon diagram representation of ET3i in complex with the 2A9 anti–C1 domain Fab antibody fragment (Protein Data Bank identifier [PDBID#] 7K66) (pink, porcine A1 domain; dark magenta, porcine A3 domain; dark teal, human A2, C1, and C2 domains; orange, 2A9 Fab light chain [LC]; green, 2A9 Fab heavy chain [HC]). (B) Direct molecular contacts between the 2A9 antibody and the C1 domain 2065-2070 loop. (C) Direct molecular contacts between the 2A9 antibody and the C1 domain 2050-2056 loop. (D) Surface contacts between the 2A9 antibody and the A3 domain 1743-1748 loop and the second N-acetylglucosamine (NAG) residue N-linked to Asn1810.
The structure of the ET3i fVIII/anti–C1 domain 2A9 Fab fragment. (A) Ribbon diagram representation of ET3i in complex with the 2A9 anti–C1 domain Fab antibody fragment (Protein Data Bank identifier [PDBID#] 7K66) (pink, porcine A1 domain; dark magenta, porcine A3 domain; dark teal, human A2, C1, and C2 domains; orange, 2A9 Fab light chain [LC]; green, 2A9 Fab heavy chain [HC]). (B) Direct molecular contacts between the 2A9 antibody and the C1 domain 2065-2070 loop. (C) Direct molecular contacts between the 2A9 antibody and the C1 domain 2050-2056 loop. (D) Surface contacts between the 2A9 antibody and the A3 domain 1743-1748 loop and the second N-acetylglucosamine (NAG) residue N-linked to Asn1810.
Conformational changes of the fVIII C2 domain
The ET3i:2A9 structure reveals a novel conformation for the C2 domain relative to the remaining ET3i structure in comparison with the unbound ET3i structure (Figure 2A).14 When aligned, the C2 domain of ET3i:2A9 is translocated forward by ∼20 Å, indicating that it has significant conformational flexibility whereas the C1 domain is locked in place relative to the A domains. Despite this large shift, the membrane-binding surface of the C2 domain still resides on the same plane as that for the C1 domain, however, this new conformation may have an impact on fIXa binding. This new conformation of fVIII raises the question of how anti-C1 inhibitors like 2A9 affect the conformation of C2 and immunogenicity of known C2 epitopes. Previous studies have demonstrated that hemophilia A patients with inhibitors possess a polyclonal antibody response to fVIII with pathogenic epitopes that recognize the A2, C1, and C2 domains.23-26 This C2 domain rearrangement is distinct from other fVIII C2 structures deposited, including the rotated conformation of C2 in the recently reported 3.2-Å X-ray structure of ET3i (supplemental Figure 2).14,21,22 In the presence of group A anti–C1 domain antibodies, an additional HDX exchange pattern is commonly observed for the 2129-2136 region.7 With this loop on the opposing side of the C1 domain that makes direct contact with the C2 domain, it is possible that this novel conformation of the C2 domain causes this HDX protection pattern and is present for other group A antibodies. Alternatively, the 2A9 epitope consists of residues that flank this loop (Thr2122, Arg2150), which may lower the conformational flexibility that is concomitant with increased HDX behavior. The new C2 domain conformation has more contacts with the C1 domain and less contacts with the A1 domain (supplemental Figure 3).
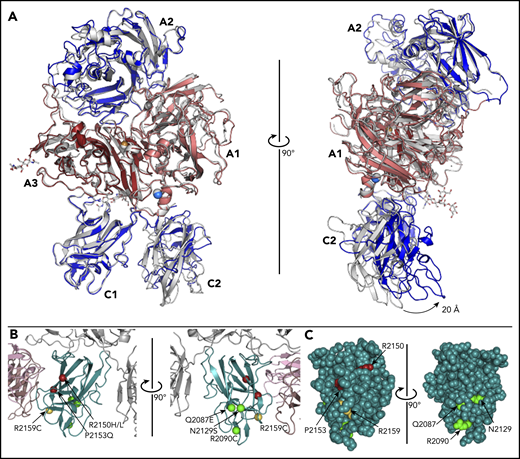
Conformational changes of the ET3i C2 domain and hemophilia A–associated mutations that decrease VWF binding. (A) Structural superposition of the unbound ET3i structure with ET3i in the 2A9 antibody complex. Arrow indicates swivel-like movement of the C2 domain (light gray, ET3i alone structure) (PDBID#, 6MF0). (B) Ribbon diagram representation of hemophilia A–associated mutations known to decrease VWF binding, represented by spheres (yellow, adjacent to 2A9 epitope; red, proximal to 2A9 epitope; green, opposing face of 2A9 epitope). (C) Van der Waals sphere representation of the fVIII C1 domain, highlighting the solvent exposure of hemophilia A–associated mutations.
Conformational changes of the ET3i C2 domain and hemophilia A–associated mutations that decrease VWF binding. (A) Structural superposition of the unbound ET3i structure with ET3i in the 2A9 antibody complex. Arrow indicates swivel-like movement of the C2 domain (light gray, ET3i alone structure) (PDBID#, 6MF0). (B) Ribbon diagram representation of hemophilia A–associated mutations known to decrease VWF binding, represented by spheres (yellow, adjacent to 2A9 epitope; red, proximal to 2A9 epitope; green, opposing face of 2A9 epitope). (C) Van der Waals sphere representation of the fVIII C1 domain, highlighting the solvent exposure of hemophilia A–associated mutations.
Hemophilia A–associated mutations that disrupt VWF binding
We searched the Centers for Disease Control and Prevention (CDC) database for hemophilia A–associated mutations that are surface exposed and/or cause impaired VWF binding to visualize surface-exposed residues that, when mutated, result in hemophilia A (Figure 2B).27-31 Although some of the mutations associated with impaired VWF binding are proximal to the 2A9 epitope (Arg2150His/Leu, Pro2153Gln), others are either adjacent (Arg2159Cys) or on the opposing face of C1 (Gln2087Glu, Arg2090Cys, Asn2129Ser). Our structure suggests that hemophilia A–associated mutations in this region may disrupt fVIII stability by disrupting VWF binding (Figure 2C).
This study reports the first structural characterization of the anti–C1 domain inhibitor response associated with hemophilia A treatment. Although both group A and B anti–C1 domain inhibitors have been shown to be pathogenic, group A only modestly inhibits fVIII activity. Batsuli et al recently demonstrated that pathogenic anti-C1 mAbs, such as 2A9, compete with VWF for binding, which increased clearance of fVIII:mAb complexes in fVIII−/− mice but not in fVIII−/−/VWF−/− mice.8 Moreover, Przeradzka et al recently showed that an fVIII F2068A mutant lowered both VWF and fIXa binding and decreased fXa generation by ∼50%.32
Select group A and all group B inhibitors block cellular uptake by human monocyte-derived dendritic cells,7 which is consistent with mutational data indicating that epitopes in this region modulate cellular uptake by antigen-presenting cells.26,33 Although 2A9 modestly blocks cellular uptake by human monocyte-derived dendritic cells, its epitope overlaps with the group AB inhibitor K33, which strongly disrupts uptake. Blocking this immunodominant epitope may suppress the immune response to fVIII, but it may also block cellular uptake by tolerogenic dendritic cells, which potentially inhibits a CD4+ T-cell–mediated antigen-specific suppression of the immune response to fVIII.34 This is consistent with a recent study showing that the 2A9 epitope is more prevalent in acquired hemophilia A patients than in congenital hemophilia A patients with inhibitor.9 Nevertheless, given that anti–C1 domain antibodies commonly block uptake of fVIII by antigen-presenting cells, understanding the immunogenicity of this region could lead to alterations in the sequence to suppress the immunogenicity of next-generation fVIII therapeutics.
The data reported in this article have been deposited in the Protein Data Bank (accession number 7K66).
Requests for data may be e-mailed to the corresponding author.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Rachel Werther and Barry Stoddard from the Fred Hutchinson Cancer Research Center for assistance with data processing at Advanced Light Source Berkeley Center for Structural Biology (ALS BCSB) beamline 5.0.1.
The Berkeley Center for Structural Biology was supported in part by the Howard Hughes Medical Institute. The Advanced Light Source is a Department of Energy Office of Science User Facility under contract no. DE-AC02-05CH11231. The Pilatus detector on 5.0.1. was funded under National Institutes of Health (NIH) grant S10OD021832 from the NIH Office of the Director. The ALS-ENABLE beamlines were supported in part by NIH National Institute of General Medical Sciences grant P30 GM124169. This work was supported by the Dreyfus Foundation (Henry Dreyfus Teacher-Scholar Award), the National Science Foundation (MRI 1429164), and the NIH National Heart, Lung, and Blood Institute (award numbers R15HL103518 and U54HL141981 [P.C.S.] and award numbers R44HL117511, R44HL110448, U54HL112309, and U54HL141981 [C.B.D., H.T.S., and P.L.]).
Authorship
Contribution: J.S.G., L.J., and K.C.C. planned experiments, performed experiments, analyzed data, and assisted in writing the manuscript; S.C.P., C.S.G., and I.W.S. performed experiments and analyzed data; H.T.S., C.B.D., and P.L. developed expression and purification procedures for ET3i and 2A9; and P.C.S. planned experiments, analyzed data, and wrote the manuscript.
Conflict-of-interest disclosure: P.L. is the inventor on a patent application describing ET3i and is an inventor on patents owned by Emory University claiming compositions of matter that include modified fVIII proteins with reduced reactivity with anti-fVIII antibodies. C.B.D., P.L., and H.T.S. are cofounders of Expression Therapeutics and own equity in the company. Expression Therapeutics owns the intellectual property associated with ET3i. The terms of this arrangement have been reviewed and approved by Emory University in accordance with its conflict-of-interest policies. The remaining authors declare no competing financial interests.
Correspondence: P. Clint Spiegel Jr, Western Washington University, 516 High St, MS 9150, Bellingham, WA 98225; e-mail: paul.spiegel@wwu.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal