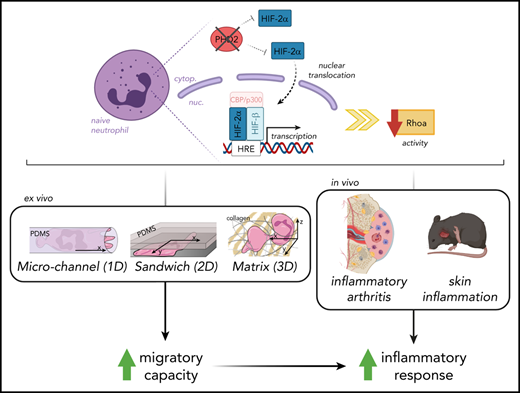
Key Points
Hypoxia pathway proteins control neutrophil motility exclusively in highly restricted environments.
PHD2-HIF2α-RhoA is a novel axis promoting movement in a chemotaxis-independent manner.

Abstract
Orchestrated recruitment of neutrophils to inflamed tissue is essential during the initiation of inflammation. Inflamed areas are usually hypoxic, and adaptation to reduced oxygen pressure is typically mediated by hypoxia pathway proteins. However, it remains unclear how these factors influence the migration of neutrophils to and at the site of inflammation during their transmigration through the blood-endothelial cell barrier, as well as their motility in the interstitial space. Here, we reveal that activation of hypoxia-inducible factor 2 (HIF2α) as a result of a deficiency in HIF prolyl hydroxylase domain protein 2 (PHD2) boosts neutrophil migration specifically through highly confined microenvironments. In vivo, the increased migratory capacity of PHD2-deficient neutrophils resulted in massive tissue accumulation in models of acute local inflammation. Using systematic RNA sequencing analyses and mechanistic approaches, we identified RhoA, a cytoskeleton organizer, as the central downstream factor that mediates HIF2α-dependent neutrophil motility. Thus, we propose that the novel PHD2-HIF2α-RhoA axis is vital to the initial stages of inflammation because it promotes neutrophil movement through highly confined tissue landscapes.
Introduction
In the innate immune response, neutrophils represent the first line of protection against infections, extravasating quickly from the circulation to inflamed tissues for fast pathogen elimination. This process necessitates transit from the oxygen-deprived bone marrow via the circulatory system to the inflammation site, which is typically hypoxic as a result of vasculature damage and/or high metabolic demand of pathogens and host cells.1
Under hypoxic conditions, the transcription factor hypoxia-inducible factor 1 (HIF1α) and its isoform, HIF2α, are key elements that control immune cell metabolism and function.2-7 Importantly, HIF activity is controlled by a class of oxygen sensors known as the HIF prolyl hydroxylase domain enzymes (PHD1-3) (for reviews, see Sormendi and Wielockx8 and Watts and Walmsley9 ). When oxygen levels decrease, PHDs get inactivated, which results in HIFα stabilization and transcription of relevant target genes. Interestingly, HIF1α deficiency results in subdued inflammation,2,3 whereas, inversely, PHD inactivation and/or HIFα stabilization leads to enhanced neutrophil survival,4,10 chemotaxis, and degranulation (for a review, see Lodge et al11 ). Although both HIFα subunits have overlapping activities, unique roles for HIF2α, including in neutrophil function, have been reported.4-6
Over the past decade, several mechanisms have been shown to participate in the multistep recruitment of neutrophils from the circulation to sites of infection or inflammation.12-14 The recruitment process requires cell plasticity, because cells deform as they move through the blood-endothelial cell barrier and the confined areas of interstitial tissues. Leukocyte migration through these microenvironments is orchestrated by actin polymerization regulators, such as the Rho GTPases RhoA, Cdc42, and Rac1.15-18 In this context, HIF1α expression has been suggested to modulate functional changes in the cytoskeleton and metabolic reprogramming.19-22 Importantly, disruption of mechanisms that control neutrophil infiltration in tissues is associated with sepsis, a life-threatening condition with multiorgan failure and 1 of the leading causes of death in the intensive care unit.23 Conversely, no effective therapeutic strategy is available for mitigating an uncontrolled neutrophilic inflammatory response.
In this study, we specifically addressed the effects of permanent PHD2 deficiency on the motility of neutrophils, including their recruitment during sterile and localized inflammation. Using ex vivo and in vivo imaging in a variety of highly confined microenvironments, we demonstrate for the first time that HIF2α overactivation enhances the migratory capacity of neutrophils in a chemotaxis-independent manner. Through whole-transcriptome analysis and combined migratory regulation, we describe a role for the PHD2-HIF2α-RhoA axis in the prompt initiation of the innate immune response.
Materials and methods
Mice
All mouse strains were housed under specific pathogen–free conditions. Experiments were performed with male and female mice at the age of 8 to 12 weeks. Vav:cre-PHD2f/f (conditional knockout [cKO] P2) and Vav:cre-PHD2/HIF2ff/ff (cKO P2H2) mouse lines were created, using PHD2f/f,24 Vav:cre25 (generous gift from Thomas Graf), and/or HIF2αf/f mice.26 All offspring were born in normal Mendelian ratios, and individual floxed lines have been previously backcrossed to C57BL/6J mice ≥9 times. Wild-type (WT) controls in all experiments were Cre− littermates without any chimerism (partial deletion of floxed genes in early blastomeres).25 Mice were genotyped using the primers described in supplemental Table 1 (available on the Blood Web site), and knockdown efficiency was confirmed via quantitative reverse transcription polymerase chain reaction on isolated neutrophils (supplemental Figure 1A,C) and/or genomic polymerase chain reaction on ear biopsies.27 KRN T-cell receptor transgenic mice were intercrossed with NOD Shilt/J mice (Charles River Laboratories, Calco, Italy) to generate K/BxN mice, as described previously.28 A detailed description of the inflammation models can be found in the supplemental Data. Breeding of all mouse lines and animal experiments were in accordance with the local guidelines on animal welfare and were approved by the Landesdirektion Sachsen (Dresden, Germany).
Results
PHD2-deficient neutrophils display enhanced migration in highly confined environments in an HIF2α-dependent manner
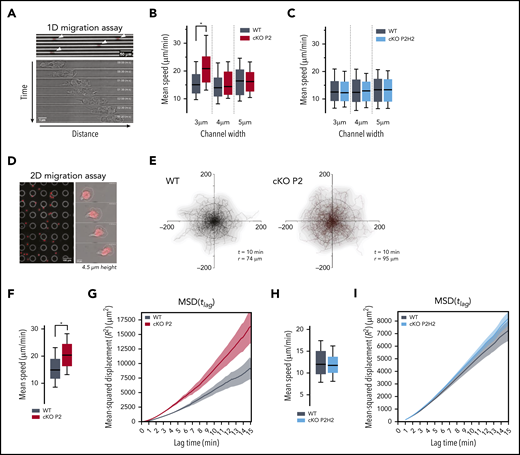
Although changes in the hypoxia pathway are involved in multiple stages of the inflammatory response, details on how the PHD/HIF axis governs neutrophil migration remain elusive. Given that PHD2 is a central regulator of the hypoxia response, we studied the motility of PHD2-deficient bone marrow–derived neutrophils (BMDNs) isolated from cKO P2 mice (supplemental Figure 1A). Initially, 1-dimensional (1D) migration assays in polydimethylsiloxane microchannel devices with different levels of constriction (channel widths of 3, 4, or 5 µm) were used to characterize the migratory capacity of individual neutrophils (Figure 1A).17,18,29-32 Interestingly, cKO P2 neutrophils moved significantly faster than did their WT counterparts, but only in the most confined channels (Figure 1B; supplemental Figure 1B). To identify downstream effectors of this phenotype, we evaluated the contributions of HIF2α in cKO P2H2 neutrophils compared with their littermate controls (supplemental Figure 1C). HIF2α has been shown to be a central regulator of neutrophil function and inflammation,4,5 and we identified it as an excellent target of PHD2 in different cell lineages in vivo.33,34 Interestingly, cKO P2H2 neutrophils did not show any differences in speed at any of the degrees of confinement tested (Figure 1C). These data strongly suggest that enhanced HIF2α activation regulates neutrophil motion in very confined microenvironments.

Neutrophils deficient for PHD2 display enhanced motility in highly confined 1D and 2D microenvironments in an HIF2α-dependent manner. (A) Example of low-resolution (original magnification, ×10) imaging of WT neutrophils migrating in 1D-microchannels (3 µm width) (upper panel). Arrowheads indicate cell position. Nucleus (red) was labeled with Hoechst. Bright-field high-resolution (original magnification, ×63) image of a neutrophil migrating in a microchannel (lower panel). (B-C) Average speed of neutrophils migrating through microchannels with a width of 3, 4, or 5 µm. At least 89 cells for the WT/cKOP2 group and 127 cells for the WT/cKOP2H2 group were measured. (D) Example of low-resolution (original magnification ×10) imaging of WT neutrophils migrating in 2D-confined devices (4.5 µm width) (left panel). Higher resolution (original magnification, ×40) image of a single neutrophil migrating in a microchannel (right panel). Nucleus (red) was labeled with Hoechst. (E) Representative neutrophil tracks during random 2D migration. t indicates the period of the displayed tracking, and r represents the mean displacement ratio in this period. (F,H) Quantification of mean speed of single neutrophil trajectories migrating in 2D confined microdevices. Panel F corresponds to tracks in panel E. Data are represented as box plots (+ median), and whiskers range from the 10th percentile to the 90th percentile. (G,I) Mean square displacement of neutrophils analyzed in panels F and H, respectively. All graphs are a representative result of ≥3 independent experiments. *P < .05, Mann-Whitney U test.
Neutrophils deficient for PHD2 display enhanced motility in highly confined 1D and 2D microenvironments in an HIF2α-dependent manner. (A) Example of low-resolution (original magnification, ×10) imaging of WT neutrophils migrating in 1D-microchannels (3 µm width) (upper panel). Arrowheads indicate cell position. Nucleus (red) was labeled with Hoechst. Bright-field high-resolution (original magnification, ×63) image of a neutrophil migrating in a microchannel (lower panel). (B-C) Average speed of neutrophils migrating through microchannels with a width of 3, 4, or 5 µm. At least 89 cells for the WT/cKOP2 group and 127 cells for the WT/cKOP2H2 group were measured. (D) Example of low-resolution (original magnification ×10) imaging of WT neutrophils migrating in 2D-confined devices (4.5 µm width) (left panel). Higher resolution (original magnification, ×40) image of a single neutrophil migrating in a microchannel (right panel). Nucleus (red) was labeled with Hoechst. (E) Representative neutrophil tracks during random 2D migration. t indicates the period of the displayed tracking, and r represents the mean displacement ratio in this period. (F,H) Quantification of mean speed of single neutrophil trajectories migrating in 2D confined microdevices. Panel F corresponds to tracks in panel E. Data are represented as box plots (+ median), and whiskers range from the 10th percentile to the 90th percentile. (G,I) Mean square displacement of neutrophils analyzed in panels F and H, respectively. All graphs are a representative result of ≥3 independent experiments. *P < .05, Mann-Whitney U test.
We extended our analysis to evaluate neutrophil migration in a 2-dimensional (2D) confined microenvironment (4.5 µm height) (Figure 1D). Similar to the results obtained in the 1D migration assay, neutrophils from cKO P2 mice showed increased motility compared with their WT counterparts, as evidenced by longer trajectories of equivalent durations (Figure 1E), greater speed (Figure 1F), and higher mean square displacement (MSD) values (Figure 1G). On the other hand, under identical conditions, cKO P2H2 neutrophils did not show any difference in speed or MSD values compared with their WT counterparts (Figure 1H-I). Interestingly, cell migration in a nonconfining 2D chamber (12 µm height) did not show any difference in speed, trajectories, or MSD value (supplemental Figure 1D-F). Thus, these data indicate that the PHD2-HIF2α pathway regulates cell migration by facilitating mobility strictly in confined spaces.
PHD2-deficient neutrophils display enhanced nondirected motility in complex environments
We used 3-dimensional (3D) collagen matrices to confirm the role of PHD2 in neutrophil migration in a microenvironment of collagen fibers of different pore sizes, mimicking the movement of the immune cell once it arrives in the interstitial tissue after extravasation. Therefore, migration of freshly isolated BMDNs from cKO P2 mice and WT littermates was compared in dense 3D collagen gels (4 mg/mL) (Figure 2A) during which cKO P2 neutrophils showed greater motility, as evidenced by a higher displacement radius (Figure 2A). Detailed analysis of these random trajectories showed that cKO P2 neutrophils displayed greater speed and MSD values compared with WT cells (Figure 2B-C). Interestingly, this difference was completely lost in less dense collagen gels (2 mg/mL) (Figure 2B; supplemental Figure 2A).
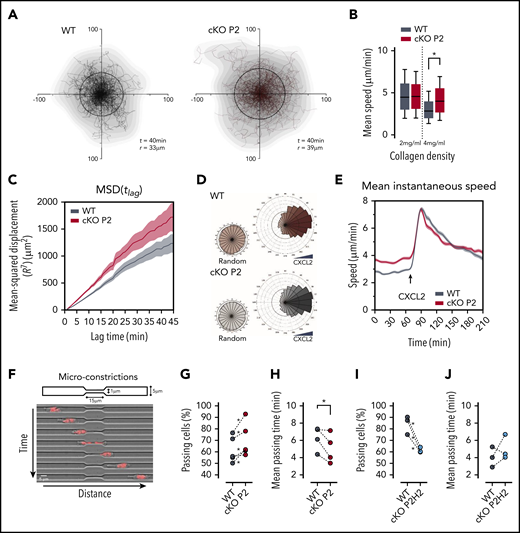
cKO P2 neutrophils display enhanced motility in highly confined 3D matrices and microconstrictions. (A) Cell trajectories of neutrophils migrating in a dense collagen gel (4 mg/mL). t indicates the period of the displayed tracking, and r is the mean displacement ratio in this period. (B) Representation of the mean speed of neutrophils migrating in a 3D matrix at different collagen concentrations (2 and 4 mg/mL). At least 157 cells for the WT group and 277 cells for the cKO P2 group were measured. Data are represented as box plots (+median), and whiskers range from the 10th percentile to the 90th percentile. Graphs are representative of ≥3 independent experiments. (C) MSD of neutrophils migrating in a 3D collagen matrix (4 mg/mL). The plot corresponds to analysis of trajectories shown in panel A. (D) Directionality of neutrophils migrating in a 4-mg/mL collagen gel prior to (random) or after CXCL2 (20 ng/mL) stimulation. At least 274 cells for the WT group and 409 cells for the cKO P2 group were traced. (E) Speed over time for cells migrating in a 4-mg/mL collagen gel. Temporal evolution of the speed before and after CXCL2 stimulation is shown. Graph corresponds to plots shown in panel D. (F) Schematic representation of the migration assay through microconstrictions. The lower panel shows an example of a cell migrating through a 1 µm × 15 μm section constriction. (G,I) Proportion of neutrophils that were able to pass through a constriction. *P < .05 in 4 of 5 independent experiments (G) and in all 3 independent experiments (I). (H,J) Average time needed by neutrophils to pass through the constriction. The statistical significance of the different experiments was defined as described in "Materials and methods." *P < .05.
cKO P2 neutrophils display enhanced motility in highly confined 3D matrices and microconstrictions. (A) Cell trajectories of neutrophils migrating in a dense collagen gel (4 mg/mL). t indicates the period of the displayed tracking, and r is the mean displacement ratio in this period. (B) Representation of the mean speed of neutrophils migrating in a 3D matrix at different collagen concentrations (2 and 4 mg/mL). At least 157 cells for the WT group and 277 cells for the cKO P2 group were measured. Data are represented as box plots (+median), and whiskers range from the 10th percentile to the 90th percentile. Graphs are representative of ≥3 independent experiments. (C) MSD of neutrophils migrating in a 3D collagen matrix (4 mg/mL). The plot corresponds to analysis of trajectories shown in panel A. (D) Directionality of neutrophils migrating in a 4-mg/mL collagen gel prior to (random) or after CXCL2 (20 ng/mL) stimulation. At least 274 cells for the WT group and 409 cells for the cKO P2 group were traced. (E) Speed over time for cells migrating in a 4-mg/mL collagen gel. Temporal evolution of the speed before and after CXCL2 stimulation is shown. Graph corresponds to plots shown in panel D. (F) Schematic representation of the migration assay through microconstrictions. The lower panel shows an example of a cell migrating through a 1 µm × 15 μm section constriction. (G,I) Proportion of neutrophils that were able to pass through a constriction. *P < .05 in 4 of 5 independent experiments (G) and in all 3 independent experiments (I). (H,J) Average time needed by neutrophils to pass through the constriction. The statistical significance of the different experiments was defined as described in "Materials and methods." *P < .05.
We verified whether this motility phenotype was dependent on HIF2α by assessing the migration of cKO P2H2 neutrophils. Surprisingly, they showed enhanced motility in 2 mg/mL and 4 mg/mL collagen gels relative to their WT counterparts (supplemental Figure 2B).
Because cKO P2H2 neutrophils showed a migratory behavior similar to that of their WT counterparts in 1D and 2D confined migration assays, this is suggestive of an effect that is dependent on the collagen microenvironment. Therefore, the enhanced migration of PHD2-deficient neutrophils in collagen gels cannot be unambiguously linked to HIF2α.
Because it has been suggested that silencing of PHD2 in neutrophils leads to their enhanced chemotaxis,35 we assessed this effect in our complex 3D collagen matrix setup using CXCL2 as chemoattractant. Directionality of cKO P2 neutrophils toward CXCL2 remained unaltered (Figure 2D). In addition, speed kinematics upon chemokine treatment were similar in PHD2-deficient and WT neutrophils (Figure 2E). Equivalent results were observed in cKO P2H2 neutrophils (supplemental Figure 2C). These results show that PHD2 deficiency does not affect CXCL2-induced neutrophil chemotactic capacity and indicate that enhanced motility in cKO P2 neutrophils is restricted to spontaneous migration.
The migratory capacity of several cell types in complex microenvironments is highly dependent on their capacity to deform when encountering narrow pores.36,37 Therefore, we evaluated whether cKO P2 neutrophils can overcome severely constricted spaces of only 1 µm width (Figure 2G).17,38 Remarkably, PHD2-deficient neutrophils showed an enhanced preference to pass through these constrictions (Figure 2H) and were also faster compared with WT neutrophils (Figure 2I). Interestingly, under these conditions, cKO P2H2 neutrophils showed reduced migration and similar migration kinetics as WT cells (Figure 2J-K), suggesting a PHD2/HIF2α-dependent axis in migration through extremely narrow constrictions.
Next, we studied whether the ability of cKO P2 neutrophils to pass through small confinements is related to changes in their deformability when an external force is applied. For this, we first analyzed neutrophil deformability using real-time fluorescence and deformability cytometry (supplemental Data), which can extract the stiffness of cells (Young’s modulus) in high throughput without contact at millisecond (ms) timescales.39,40 We used steady-state BMDNs, phorbol 12-myristate 13-acetate (PMA)-activated BMDNs, and peripheral blood neutrophils isolated 6 hours after thioglycolate-induced peritonitis. However, no difference was observed between cKO P2 and WT neutrophils under any of the conditions tested (supplemental Figure 2D). Likewise, a microcapillary microcirculation mimetic assay41,42 using peritonitis neutrophils did not show any difference in their ability to passively navigate through multiple constrictions at high speed (supplemental Figure 2E). Taken together, these assays strongly suggest that loss of PHD2 does not affect neutrophil deformability under externally applied stress without confinement.
PHD2-deficient neutrophils extravasate faster in vivo and accumulate in inflamed tissue
Based on the enhanced ability of PHD2-deficient neutrophils to overcome very small constrictions, we decided to study the behavior of these cells in vivo, specifically in a more complex setting of sterile skin inflammation. Earlobes from cKO P2 and WT littermate mice that did not display any difference in the total numbers of hematopoietic stem cells, myeloid progenitors, or mature neutrophils (supplemental Figure 3A) were ectopically treated with PMA, and the recruitment of Ly6G+ cells was visualized using intravital 2-photon microscopy (Figure 3A). In line with our previous experiments, we found that PHD2-deficient neutrophils were able to extravasate ∼30% faster from the vessel into the ear tissue than were their WT counterparts (Figure 3B-C). Furthermore, the cumulative effect of faster neutrophil extravasation time resulted in an anticipated increase in Gr1+ cells in the inflamed cKO P2 ear compared with that in WT littermates at 24 hours after PMA treatment (Figure 3D). Conversely, but consistently, this difference in migration was abolished in cKO P2H2 mice (Figure 3E), further confirming a role for HIF2α activity in driving the increased migration capacity of these neutrophils.
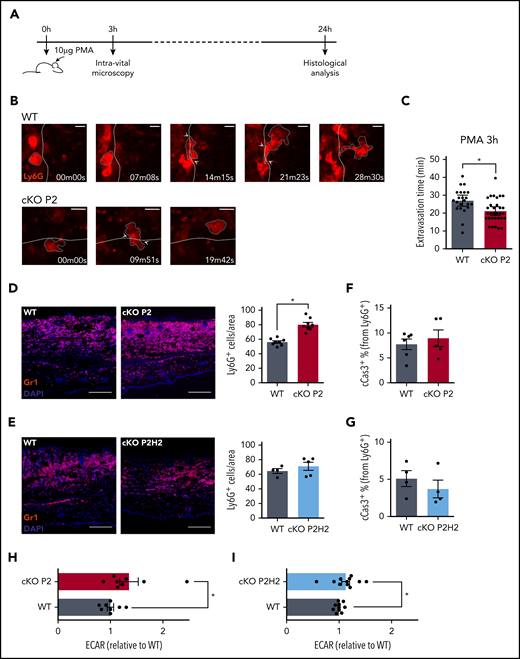
Loss of PHD2 in neutrophils enhances the speed of transendothelial migration in a mouse model of acute skin inflammation. Schematic representation of the acute skin inflammation model (A), including time points for the intravital microscopy analysis (B) and final histological analyses (D-G). (B-C) Intravital imaging of representative neutrophils using 2-photon microscopy, 3 hours after PMA was applied to the ear (WT vs cKO P2). Time point 00m00s shows Ly6G+ neutrophils (+ white dashed lines surrounding the red cell) that had stopped rolling/moving at the blood-endothelial barrier (= white dashed lines) before diapedesis and migration into the inflamed tissue. The last frames represent the first time point that the individual neutrophil had completely left the bloodstream (scale bars, 10 μm). (C) The average time necessary for individual neutrophils to complete the diapedesis. Total amount of cells were collected from 3 individual mice per genotype. Representative immunofluorescent images of Gr1+ neutrophils on ear sections 24 hours after PMA application (D-E, scale bars, 50 μm) and quantification of the total number of cells per area or fraction of apoptotic neutrophils (cCas3+) (F-G). Each data point represents an average amount based on ≥6 images per individual mouse. (H-I) Extracellular acidification rate (ECAR) measurements from steady-state neutrophils immediately after negative selection. Data points represent individual mice from ≥3 experiments - normalized values against WT control. Data are mean ± standard error of the mean. *P < .05, Mann-Whitney U test.
Loss of PHD2 in neutrophils enhances the speed of transendothelial migration in a mouse model of acute skin inflammation. Schematic representation of the acute skin inflammation model (A), including time points for the intravital microscopy analysis (B) and final histological analyses (D-G). (B-C) Intravital imaging of representative neutrophils using 2-photon microscopy, 3 hours after PMA was applied to the ear (WT vs cKO P2). Time point 00m00s shows Ly6G+ neutrophils (+ white dashed lines surrounding the red cell) that had stopped rolling/moving at the blood-endothelial barrier (= white dashed lines) before diapedesis and migration into the inflamed tissue. The last frames represent the first time point that the individual neutrophil had completely left the bloodstream (scale bars, 10 μm). (C) The average time necessary for individual neutrophils to complete the diapedesis. Total amount of cells were collected from 3 individual mice per genotype. Representative immunofluorescent images of Gr1+ neutrophils on ear sections 24 hours after PMA application (D-E, scale bars, 50 μm) and quantification of the total number of cells per area or fraction of apoptotic neutrophils (cCas3+) (F-G). Each data point represents an average amount based on ≥6 images per individual mouse. (H-I) Extracellular acidification rate (ECAR) measurements from steady-state neutrophils immediately after negative selection. Data points represent individual mice from ≥3 experiments - normalized values against WT control. Data are mean ± standard error of the mean. *P < .05, Mann-Whitney U test.
Because previous studies have described PHD2-related improved survival of neutrophils during inflammation,4,43 we evaluated the level of apoptotic cells in ears that were treated with PMA for 24 hours, but we did not find any difference in cleaved caspase-3+ cell numbers (cCas3+) between the different genotypes (Figure 3F-G; supplemental Figure 3B-C). This was also confirmed in different ex vivo setups; no difference was observed in the amount of apoptotic neutrophils after serum starvation or subsequent efferocytosis by WT or cKO P2 bone marrow–derived macrophages (supplemental Figure 3D-E). Additionally, because recent work has associated PHD2 with enhanced neutrophil glycolysis and their recruitment to sites of inflammation,35 we assessed the glycolytic capacity of BMDNs from cKO P2 mice, cKO P2H2 mice, and their respective WT counterparts by measuring the extracellular acidification rate. In line with previous reports, PHD2-deficient neutrophils appeared to be significantly more glycolytic than their respective WT counterparts (Figure 3H). However, neutrophils lacking PHD2 and HIF2α also showed significantly higher glycolysis (Figure 3I). Taken together, although HIF2α directly controls the migration speed of neutrophils in confined spaces and inflamed tissues, this effect is independent of their survival or glycolytic activity.
HIF2α stabilization upon loss of PHD2 affects cytoskeletal gene expression profiles
It is well accepted that the functionality of innate immune cells varies depending on the lipid-type composition of their cytoplasmic membrane.44,45 Therefore, we evaluated whether an altered membrane lipid composition of cKO P2 neutrophils could account for their different migratory abilities by performing high-throughput lipidomic analysis of freshly isolated BMDNs (supplemental Data). However, because there were not any significant alterations between cKO P2 and WT BMDNs (supplemental Figure 2D; supplemental Table 3), it appears unlikely that differences in the lipid composition are directly responsible for the dramatic difference in the migratory capacity of cKO P2 neutrophils.
Next, to further characterize the molecular underpinnings of the HIF2α-driven neutrophil migration phenotype, we used next-generation sequencing to analyze the steady-state transcriptomes of BMDNs derived from cKO P2 and cKO P2H2 mice and compare it with those of their respective WT counterparts (Figure 4A). Gene signatures of various lineages were evaluated using gene-set enrichment analysis (GSEA), as described previously.46-48 In line with our in vivo cCas3+ results, we did not detect any significant apoptosis signatures among any of the genotypes (Figure 4B), and next-generation sequencing confirmed a significant enrichment of glycolysis/gluconeogenesis-related genes in cKO P2 and cKO P2H2 BMDNs (Figure 4C). Strikingly, steady-state BMDNs lacking PHD2, with or without HIF2α, displayed a significant reduction in genes related to the innate immune response but not the chemokine-signaling pathway (Figure 4D). Together, these observations suggest that significant HIF2α-independent changes in the glycolytic capacity and immune response of PHD2-deficient neutrophils can likely be linked to HIF1α activity, as previously suggested.2,35
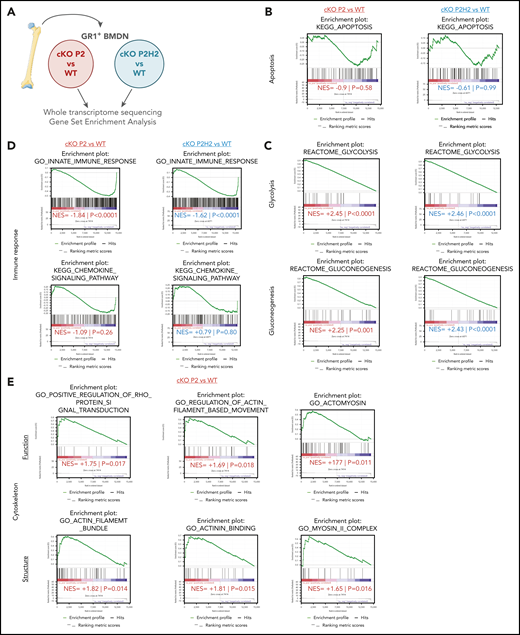
PHD2-deficient neutrophils present a distinct signature associated to cytoskeleton dynamics. (A) Schematic overview of the deep-sequencing approach, comparing RNA sequencing data from cKO P2 and cKO P2H2 mice, as well as their respective WT littermates. Apoptosis signatures in both transgenic lines are not changed significantly compared with their WT littermates (B), whereas glycolysis/gluconeogenesis signatures are highly correlated with cKO P2 and cKO P2H2, suggesting an HIF2-independent phenotype (C). (D) cKO P2 and cKO P2H2 mice display a negative correlation, with signatures related to the innate immune response but not chemokine signaling. (E) A number of signatures significantly linked to functioning and structure of the cytoskeleton are found only in cKO P2 vs WT littermate neutrophils. NES, normalized enrichment score.
PHD2-deficient neutrophils present a distinct signature associated to cytoskeleton dynamics. (A) Schematic overview of the deep-sequencing approach, comparing RNA sequencing data from cKO P2 and cKO P2H2 mice, as well as their respective WT littermates. Apoptosis signatures in both transgenic lines are not changed significantly compared with their WT littermates (B), whereas glycolysis/gluconeogenesis signatures are highly correlated with cKO P2 and cKO P2H2, suggesting an HIF2-independent phenotype (C). (D) cKO P2 and cKO P2H2 mice display a negative correlation, with signatures related to the innate immune response but not chemokine signaling. (E) A number of signatures significantly linked to functioning and structure of the cytoskeleton are found only in cKO P2 vs WT littermate neutrophils. NES, normalized enrichment score.
Conversely, a number of HIF2α-dependent gene signatures associated with PHD2 deficiency were related to the function and structure of the neutrophil cytoskeleton, including Rho GTPase activity (Figure 4E). Additionally, using an integrative method, we identified a number of master regulators that could potentially control cellular cytoskeletal rearrangements through transcriptional or protein regulation (supplemental Figure 4A).
Diminished RhoA GTPase activity underlies the faster HIF2α-dependent migration of PHD2-deficient neutrophils
Small Rho GTPases (RhoA, Cdc42, and Rac) are key molecular effectors that steer cytoskeletal dynamics. Although we did not find any difference in the messenger RNA expression of any of these 3 GTPases (cKO vs WT) (supplemental Figure 4B), we identified numerous potential protein-protein associations between RhoA and/or Cdc42 (but not Rac) and 49 gene products identified in the GSEA signatures (Figure 5A). A number of them were identified as master regulators (supplemental Figure 4A) or contained putative HIF2α binding sites (supplemental Figure 4C).49
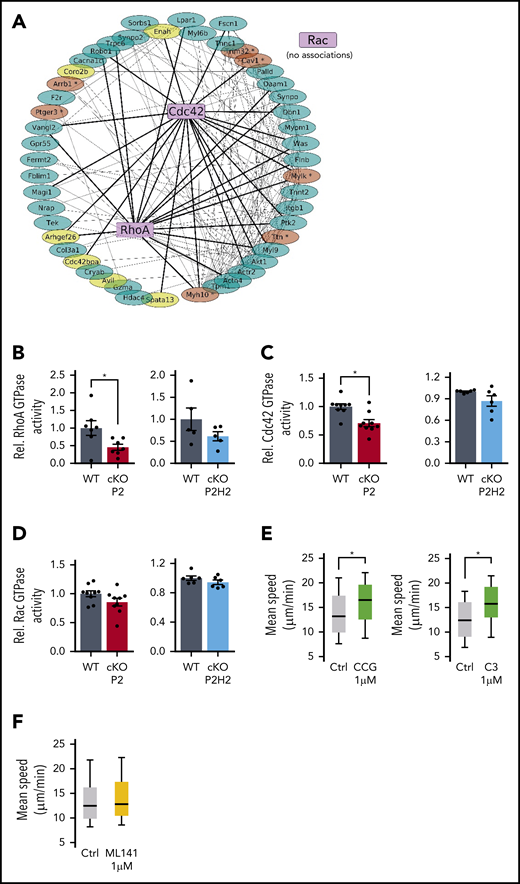
Reduced RhoA-GTPase activity in cKO P2 neutrophils leads to enhanced speed in highly confined environments. (A) The protein-protein interaction (PPI) network map was built from the 49 genes from GSEA signatures: “cytoskeleton remodeling” and the small GTPases. Bold lines are potential PPIs between RhoA or Cdc42 and GSEA signature gene products, and thin lines associates both signature genes. Genes in yellow are defined as master regulators, and genes in brown contain a putative HIF2α binding site (supplemental Figure 4A,C). (B-D) Rho GTPase activity measured in steady-state BMDNs from all different genotypes and their WT littermates. Data points represent individual mice from ≥2 experiments - normalized values against WT control. Data are mean ± standard error of the mean. Quantification of average speed of WT neutrophils treated with different inhibitors against RhoA (E) or Cdc42 inhibitor (F) in 3-µm-wide microchannels. Data are represented as box plots (+median), and whiskers range from the 10th percentile to the 90th percentile. Data points are representative of 2 independent experiments. *P < .05, Mann-Whitney U test.
Reduced RhoA-GTPase activity in cKO P2 neutrophils leads to enhanced speed in highly confined environments. (A) The protein-protein interaction (PPI) network map was built from the 49 genes from GSEA signatures: “cytoskeleton remodeling” and the small GTPases. Bold lines are potential PPIs between RhoA or Cdc42 and GSEA signature gene products, and thin lines associates both signature genes. Genes in yellow are defined as master regulators, and genes in brown contain a putative HIF2α binding site (supplemental Figure 4A,C). (B-D) Rho GTPase activity measured in steady-state BMDNs from all different genotypes and their WT littermates. Data points represent individual mice from ≥2 experiments - normalized values against WT control. Data are mean ± standard error of the mean. Quantification of average speed of WT neutrophils treated with different inhibitors against RhoA (E) or Cdc42 inhibitor (F) in 3-µm-wide microchannels. Data are represented as box plots (+median), and whiskers range from the 10th percentile to the 90th percentile. Data points are representative of 2 independent experiments. *P < .05, Mann-Whitney U test.
To substantiate this link between PHD2/HIF2α and Rho GTPases, we used an ex vivo enzymatic assay to quantify the activity of these Rho GTPases in untreated freshly isolated BMDNs from cKO P2 and P2H2 mice. Interestingly, cKO P2 neutrophils exhibited diminished RhoA and Cdc42 GTPase activity (Figure 5B-C), whereas Rac GTPase activity was comparable to that of WT neutrophils (Figure 5D). Further, cKO P2H2 neutrophils did not display any significant reduction in RhoA, Cdc42, or Rac GTPase activity, suggesting that regulation of RhoA and/or Cdc42 is dependent on the PHD2/HIF2α-axis (Figure 5B-D).
Given this reduction in RhoA and Cdc42 GTPase activity in PHD2-deficient neutrophils, we examined whether their direct inhibition in WT neutrophils can mimic the motility phenotype displayed by cKO P2 neutrophils. Therefore, we performed a series of ex vivo 1D-migration assays using the RhoA inhibitor CCG100602 and the Rho inhibitor exoenzyme C3 transferase. Although the use of low doses of CCG100602 or exoenzyme C3 transferase enhanced the speed of migrating neutrophils in 3-µm microchannels (Figure 5E), treatment of cells with a Cdc42 inhibitor (ML141) did not have any effect on the velocity of BMDNs (Figure 5F). Taken together, our data argue strongly for a PHD2/HIF2α-orchestrated regulatory loop in the RhoA GTPase activity–dependent motility of BMDNs.
The PHD2/HIF2α axis controls neutrophil accumulation in joints during severe inflammatory arthritis
To test the biological effects of the enhanced migratory capacity of PHD2-deficient neutrophils, we subjected the different mouse strains to an autoantibody-induced inflammatory arthritis model (K/BxN), which has been shown to be myeloid dependent (Figure 6A).50,51 cKO P2 mice displayed enhanced swelling of the hind limbs compared with their WT littermates (Figure 6B), and this effect was sustained throughout the first 2 weeks of the experiment. In line with our previous results, cKO P2H2 mice and their WT littermates did not display any difference in swelling (Figure 6C). To characterize the myeloid composition of the inflamed knee joints, we performed flow cytometry analysis of the synovial fluid drawn on day 5, which revealed much higher accumulation of neutrophils in cKO P2 knee joints (approximately threefold increase vs WT), along with slightly enhanced macrophages (Figure 6D). Importantly, these differences were not associated with changes in survival, because the genotypes displayed similar fractions of viable Ly6G+ cells in their synovial fluid (WT vs cKO P2: 90.8% ± 3.6% vs 89.15% ± 2.2%; mean ± standard deviation). Immunofluorescence for Gr1 on sections of knee joints (day 5) further confirmed the increased amount of neutrophils (Figure 6E). Conversely, although no difference was observed in joint swelling between cKO P2H2 mice and their WT littermates, their synovial fluid showed a slight, but significant, reduction in neutrophil numbers at day 5 compared with cKO P2 mice (Figure 6F). Thus, PHD2/HIF2α is also a central axis during the initial stages of inflammation in arthritic joints.
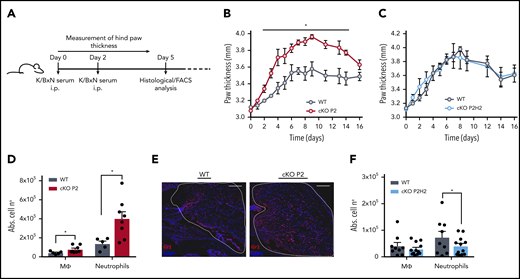
HIF-2α enhances neutrophil recruitment to the synovium in a mouse model of acute inflammatory arthritis. (A) Schematic representation of K/BxN serum-induced inflammatory arthritis. The inflammatory response was measured as paw thickness (mm) in cKO P2 mice (B) and in cKO P2H2 mice (C) vs their representative control littermates (n = 4). Data are representative of ≥6 (B) and 3 (C) individual experiments. Neutrophil recruitment was assessed via flow cytometry analysis, measured as absolute numbers (Abs. cell no) of neutrophils and macrophages in the synovial cavity in cKO P2 mice (n = 5-8) (D) and cKO P2H2 mice (F) vs WT littermates (n = 9-12). (E) Representative images of Gr1 immunofluorescence staining of the synovial cavity (dashed line) 5 days after the start of the experiment (scale bars, 50 μm). Data are represented as mean ± standard error of the mean. *P < .05, Mann-Whitney U test.
HIF-2α enhances neutrophil recruitment to the synovium in a mouse model of acute inflammatory arthritis. (A) Schematic representation of K/BxN serum-induced inflammatory arthritis. The inflammatory response was measured as paw thickness (mm) in cKO P2 mice (B) and in cKO P2H2 mice (C) vs their representative control littermates (n = 4). Data are representative of ≥6 (B) and 3 (C) individual experiments. Neutrophil recruitment was assessed via flow cytometry analysis, measured as absolute numbers (Abs. cell no) of neutrophils and macrophages in the synovial cavity in cKO P2 mice (n = 5-8) (D) and cKO P2H2 mice (F) vs WT littermates (n = 9-12). (E) Representative images of Gr1 immunofluorescence staining of the synovial cavity (dashed line) 5 days after the start of the experiment (scale bars, 50 μm). Data are represented as mean ± standard error of the mean. *P < .05, Mann-Whitney U test.
Discussion
In this study, we explored whether hypoxia pathway proteins can directly regulate neutrophil motility and revealed that sustained activation of HIF2α in mouse neutrophils due to constitutive PHD2 loss enhances neutrophil migration through very confined environments independent of chemotactic, glycolytic, or apoptotic activity. Using a combination of in vivo, ex vivo, and deep-sequencing approaches, we provide evidence that the enhanced migratory capacity of cKO P2 neutrophils relates to changes in their cytoskeleton dynamics that are mediated by a substantial reduction in RhoA GTPase activity.
Although it is generally accepted that neutrophils are the first immune cells to arrive in the tissue during inflammation, the molecular basis of neutrophil recruitment, which encompasses extravasation and interstitial migration, remains elusive. Further, neutrophil recruitment has been evaluated using a variety of migration assays in multiple studies related to the innate immune response,52-55 including in the context of hypoxia pathway proteins,2,4 but these studies call into debate the role of adhesion molecules.56,57 Here, we consistently show that neutrophils lacking PHD2 display enhanced cell motility in severely confined 1D, 2D, and 3D microenvironments, an effect that we found to be HIF2α dependent in 1D and 2D settings. At this point, it is unclear whether overactivation of HIF2α is central in neutrophil movement through very confined 3D collagen structures because cKO P2H2 neutrophils showed enhanced velocity in 3D collagen gels, but it was independent of the density. A potential explanation for this observation is that the absence of PHD2 and HIF2α affects the interaction of neutrophils with collagen fibers, exclusively altering their migration capacity independently of the confinement level. Moreover, enhanced chemokinesis of cKO P2 neutrophils did not interfere with their chemotactic capacities, because their movement toward the gradient was unidirectional and reached a maximum speed defined by the complexity of the collagen network (eg, density, pore size) and the chemokine. This demonstrates that the enhanced migratory capacity regulated by the PHD2-HIF2α axis is probably a cell-intrinsic characteristic.
An important process during neutrophil recruitment is the final and time-limiting step of transendothelial migration, which is mediated, in part, by mechanical forces generated by the migrating neutrophil itself.58-60 We reveal a central role for HIF2α in this process. Indeed, considering the narrow pores between neighboring endothelial cells during the early phase of neutrophil diapedesis,59 our results from multiple approaches reiterate 2 main observations: greater numbers of cKO P2 neutrophils pass through small constrictions with enhanced speed. Intuitively, these observations account for the shorter transendothelial migration time in a local ear inflammation model. The cumulative effects of such enhanced transmigration into inflamed tissues were observable even at later time points in 2 completely independent in vivo models (ie, inflammatory skin lesions and sterile arthritis). Indeed, it is possible that the enormous increase in cKO P2 neutrophils is positively affected by the fact that once a pore is opened, successive neutrophils are more likely to extravasate at this spot, enabling more neutrophils to enter the interstitium (skin) or the synovium (joint) of the inflamed tissue.59
Previous studies in a model of acute lung injury and different ex vivo approaches have reported enhanced glycolytic capacity of PHD2-deficient neutrophils, potentially due to HIF1α stabilization, which was associated with enhanced neutrophil recruitment to the inflammatory site.35 Here, we confirm enhanced glycolysis in cKO P2 neutrophils and show that it is independent of HIF2α, strongly suggesting that glycolytic metabolism does not underlie the migratory phenotype described in our study. The absence of differences in neutrophil apoptosis in vivo was corroborated by the RNA sequencing data from single- and double-knockout neutrophils, implying that the prolonged inflammation phenotype in cKO P2 mice probably was not due to the persistence of the neutrophils. This is in contrast to results obtained using in vitro approaches that describe reduced apoptosis in HIF2α-overexpressing neutrophils, which then resulted in delayed resolution of the inflammatory response.4 That group also reported delayed apoptosis in PHD2-deficient neutrophils and connected this to persistent inflammation.35 We believe that these discrepancies are related to differences in the experimental models used.
Although several studies have linked the hypoxia pathway to the migratory capacity of a cell, only a few have suggested a role for the PHD/HIF axis in regulating cell migration through changes in cytoskeletal function.20-22 In migrating neutrophils in vivo, dynamic polymerized actin converges at the leading edge of pseudopods, whereas stable actin with high actomyosin contractility assembles at the rear. Polarization and maintenance of this cytoskeletal asymmetry strongly rely on Rho GTPase activity.61,62 Using deep-sequencing data from neutrophils of single- and double-transgenic lines, we show that a vast number of genes associated with Rho GTPase signaling are directly or indirectly regulated by HIF2α. Interestingly, cKO P2 neutrophils displayed a significant downregulation of RhoA GTPase activity. These findings are in agreement with those reported earlier (ie, increased flux of RhoA-deficient neutrophils and aggravated tissue damage in lipopolysaccharide-induced acute lung injury).63 Our data also show that RhoA activity is not completely abolished in cKO P2 neutrophils. To mimic this condition in vitro, we used low amounts of RhoA inhibitor in the migration experiments. However, we noticed that the use of high doses of RhoA inhibitor decreased neutrophil migration under high confinement (M.D. and P.V., unpublished results). This indicates that RhoA might have a dual role in the regulation of neutrophil migration. In agreement, Jennings and colleagues found increased neutrophil motility in the absence of RhoA only upon adhesion-independent stimuli.63 Because leukocyte migration under confinement does not depend on cell adhesions,64 it is possible that the mode of motility used by neutrophils under strong compression is different from the one used in the absence of confinement, as proposed in activated dendritic cells.33
A potential explanation is that partial RhoA inhibition would primarily decrease dynamic protrusions at the cell front, which is known to restrict cell migration by competing with stable actin cables at the cell rear.18 Such a mechanism has been already proposed to be required to sustain cell polarity during neutrophil migration.65
Alternatively, the HIF2α axis could be directly involved in the induction of cell contractility, which promotes neutrophil and dendritic cell migration under strong confinement.17,32 However, further efforts are required to identify the specific molecular mechanism. However, it cannot be excluded whether there is also a relative contribution of HIF1α activation to the motility of the PHD2-deficient neutrophils in confined environments other than via enhanced glycolysis. In that respect, HIF1 has been associated with enhanced cell motility and migratory capacity that result from the accumulation of F-actin22 or even colocalization of F-actin with pyruvate kinase muscle 2 in filopodia.19
In summary, our results demonstrate that HIF2α-activation, resulting from constitutive loss of PHD-2, enhances the motility of neutrophils in highly confined surroundings, also during inflammation. Importantly, this phenotype is independent of chemotaxis signaling, glycolysis, or apoptosis. Mechanistically, the reduction in RhoA GTPase activity enhances the motility of PHD2-deficient neutrophils through very confined microenvironments. These findings highlight the potential deleterious effects of sustained HIF2α activity and may have important implications for the uncontrolled use of hypoxia-mimetic agents that are currently licensed or are in phase 2 or phase 3 clinical trials.
The RNA sequencing data reported in this article have been deposited in the Gene Expression Omnibus database (accession number GSE151703).
Data sharing requests should be sent to Pablo Vargas (pablo.vargas@curie.fr) or Ben Wielockx (ben.wielockx@tu-dresden.de).
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Silke Tulok and Anja Nobst (Core Facility Cellular Imaging/Medical Theoretical Center [CFCI-MTZ]-Dresden) and Johanna Stein for assistance, Thomas Graf (Centre for Genomic Regulation [CRG], Barcelona, Spain) for the Vav:cre mouse line, and Vasuprada Iyengar for English language and content editing.
This work was supported by grants from the Deutsche Forschungsgemeinschaft (DFG) (TRR-CRC 205 Die Nebenniere: Zentrales Relais in Gesundheit und Krankheit [A02] [B.W. and T.C.] and CRC 1181 [C7] [T.C.]), the Alexander von Humboldt Foundation (AvH Professorship to J.G.), and Fondation pour la Recherche Médicale (SPF201809007121) (P.V.). M.B. received financial support from the Association Nationale pour la Recherche (ANR) (MOTILE Project, ANR-16-CE13-0009), the Emergences Cancéropôle (SYNTEC Project), Laboratoire d’Excellence–Institut Pierre-Gilles de Gennes (LabEx-IPGG) “Investissements d’Avenir” Program (ANR-10-IDEX-0001-02 PSL and ANR-10-LABX-31). S.S. received financial support from the Dresden International Graduate School for Biomedicine and Bioengineering. B.W. was supported by the Heisenberg Program (DFG, WI3291/5-1 and 12-1). M.D. was supported by the Fondation pour la Recherche Médicale (SPF201809007121). M.B. and P.V. were funded by ATEurope and Cancéropôle Ile-de-France (IDF) (SYNTEC Project).
Authorship
Contribution: S.S. designed the study, performed the majority of experiments, analyzed data, and wrote the manuscript; M.D., I.K., K.F., and M.K. designed and performed experiments, analyzed data, and contributed to the discussions; P.J.S., G.L.L., and M.B. analyzed data and contributed to the discussions; A.S. performed deep sequencing analyses; A.P. performed lipidomics analyses; M.G. performed intravital microscopy and contributed to the discussions; A.K. and A.M. performed experiments and analyzed data; J.G. and Ü.C. provided tools and contributed to the discussions; T.C. provided tools, contributed to the discussions, and edited the manuscript; P.V. designed and supervised the ex vivo migration studies, performed experiments, analyzed data, and wrote the manuscript; and B.W. designed and supervised the overall study, analyzed data, and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
The current affiliation for I.K. is Hull York Medical School, York Biomedical Research Institute, University of York, York, United Kingdom.
The current affiliation for M.K. and J.G. is Max Planck Institute for the Science of Light and Max-Planck-Zentrum für Physik und Medizin, Erlangen, Germany.
Correspondence: Pablo Vargas, Systems Biology of Cell Polarity and Cell Division, Institut Curie (UMR 144) and Institut Pierre Gilles de Gennes, 6 rue Jean Calvin, 75005 Paris, France; e-mail: pablo.vargas@curie.fr; and Ben Wielockx, Institute of Clinical Chemistry and Laboratory Medicine, Technical University Dresden, Fetscherstr 74, 01307 Dresden, Germany; e-mail: ben.wielockx@tu-dresden.de.