Key Points
MB1-47, a novel mitochondrial uncoupler with vastly improved pharmacokinetics, shows a good therapeutic window for clinical use.
Mitochondrial uncoupling in mouse primary T-ALL and human PDXs leads to metabolic collapse and highly antileukemic effects in vivo.

Abstract
T-cell acute lymphoblastic leukemia (T-ALL) is an aggressive hematologic malignancy. Despite recent advances in treatments with intensified chemotherapy regimens, relapse rates and associated morbidities remain high. In this context, metabolic dependencies have emerged as a druggable opportunity for the treatment of leukemia. Here, we tested the antileukemic effects of MB1-47, a newly developed mitochondrial uncoupling compound. MB1-47 treatment in T-ALL cells robustly inhibited cell proliferation via both cytostatic and cytotoxic effects as a result of compromised mitochondrial energy and metabolite depletion, which severely impaired nucleotide biosynthesis. Mechanistically, acute treatment with MB1-47 in primary leukemias promoted adenosine monophosphate–activated serine/threonine protein kinase (AMPK) activation and downregulation of mammalian target of rapamycin (mTOR) signaling, stalling anabolic pathways that support leukemic cell survival. Indeed, MB1-47 treatment in mice harboring either murine NOTCH1-induced primary leukemias or human T-ALL patient-derived xenografts (PDXs) led to potent antileukemic effects with a significant extension in survival without overlapping toxicities. Overall, our findings demonstrate a critical role for mitochondrial oxidative phosphorylation in T-ALL and uncover MB1-47–driven mitochondrial uncoupling as a novel therapeutic strategy for the treatment of this disease.
Introduction
T-cell acute lymphoblastic leukemia (T-ALL) is a relatively rare lymphoid neoplasm clinically characterized by elevated white cell counts, mediastinal thymic masses, and frequent meningeal infiltration of the central nervous system.1 Management of newly diagnosed T-ALL cases is based on multiagent intensive chemotherapy regimens.2 Despite the efficacy of these regimens, 20% of pediatric and over 50% of adult T-ALL cases still show primary resistance and/or relapse3,4; the prognosis of these cases is extremely poor, with <10% 5-year survival.5 Remarkably, a high percentage of all casualties are due to toxic side effects during remission, usually from severe infections.6 Additionally, most T-ALL survivors present further serious neurocognitive and cardiovascular impairments,7,8 highlighting the need to develop safer strategies that might reduce morbidity and mortality rates.
A key feature of cancer cells is their capability to sustain chronic, uncontrolled proliferation and enhanced survival. In this context, one of the critical hallmarks of human cancer is cancer-specific metabolic rewiring, thus, selective targeting of primary cellular metabolic routes might be an attractive therapeutic option.9 Indeed, antimetabolite drugs have been used in the clinic to treat hematologic malignancies for decades and common regimens for leukemias include: purine antimetabolites, such as 6-mercaptopurine; pyrimidine antimetabolites, such as cytarabine; or antifolates, such as methotrexate.10 Notably, we recently demonstrated that targeting serine catabolism via serine hydroxymethyltransferase inhibition shows highly antileukemic effects on its own and synergizes with methotrexate, uncovering a promising novel therapeutic target for this disease.11 Unfortunately, T-ALL cells still develop mechanisms of resistance12,13 and antimetabolite drugs usually produce adverse effects in normal proliferative tissues causing severe diarrheas and/or immunosuppression.14,15 In line with this, l-asparaginase, a crucial component for the success of conventional regimens in T-ALL cases, takes advantage of a particular metabolic vulnerability in leukemic cells.16,17 This treatment shows outstanding efficacy because asparagine is required for leukemia cell growth, and T-ALL cells are highly dependent on extracellular pools of this amino acid because they express low levels of asparagine synthetase.18 However, adverse events requiring chemotherapy cessation occur in up to one-third of patients.19 Thus, designing novel molecules, with safer profiles to target metabolic routes on which leukemic cells exhibit higher dependency than normal proliferating cells, is a top priority in the field.
The detection of highly prevalent NOTCH1 activating mutations (∽60% of patients) in T-ALL20 paved the way for more personalized targeted therapy and led to the discovery of NOTCH1-signaling inhibitors, such as γ-secretase inhibitors. γ-secretase inhibitors block a critical proteolytical cleavage step required for NOTCH1 maturation and activation,21 and are currently being explored in clinical trials for T-ALL relapsed/refractory cases. However, the responses observed as a single-agent regimen have been limited due to the severe gastrointestinal toxicity22 and the acquisition of resistance mechanisms, such as the loss of phosphatase and tensin homolog (PTEN).23,24 These traits have limited the extensive use of these drugs, and current preclinical studies are focused on discovering new tumor liabilities to identify synthetic lethal interactions. Interestingly, oncogenic NOTCH1 induces metabolic stress and promotes oxidative phosphorylation (OXPHOS) in T-ALL cells.25 We previously demonstrated that the effects of NOTCH1 inhibition on central carbon metabolism are critical for its antileukemic activity in T-ALL in vivo.24 In this context, we hypothesized that uncoupler drugs that target mitochondrial OXPHOS might be potent antileukemic agents. To test this hypothesis, we examined the efficacy and metabolic effects of a novel niclosamide-based second-generation mitochondrial uncoupling drug in T-ALL cell lines, mouse primary leukemias with isogenic loss of Pten or adenosine monophosphate (AMP)-activated protein kinase (Ampk), and clinically relevant patient-derived xenografts (PDXs) in vivo.
Methods
Mice
Generation of NOTCH1-driven conditional inducible knockout leukemias was done as previously described,24 and is further detailed in supplemental Methods (available on the Blood Web site).
For survival studies, 1 × 106 mouse primary leukemia cells were transplanted from primary recipients into 6- to 8-week-old sublethally irradiated (4.5 Gy) C57BL/6 secondary recipients (Taconic Farms) by retro-orbital injection. To induce isogenic deletion of Pten or Ampka1, 2 days after leukemic cell transplantation recipient mice were treated with 3 mg of tamoxifen (Sigma) dissolved in corn oil (ACROS Organics) at 30 mg/mL via IP injection. Subsequently, 5 days posttransplantation, mice in each arm were divided randomly into 2 different groups: control groups were subsequently fed with a normal control diet (AIN-93M; Research Diets) whereas treated groups were subsequently fed with an AIN-93M diet containing MB1-47 (1000 ppm, equivalent to a dose of 100 mg/kg body weight per day). Investigators were not blinded to group allocation. Animals were monitored for signs of distress or motor function at least twice daily, until they were terminally ill, whereupon they were euthanized. All animal housing, handling, and procedures involving mice were approved by the Rutgers Institutional Animal Care and Use Committee, in accordance with all relevant ethical regulations.
Human primary xenografts
PDTALL19 and PDTALL9 samples were provided by the University of Padova. The CU-L-157 sample was provided by Columbia University. Patients were informed beforehand and signed the written consent. Samples were collected under the supervision of local institutional review boards and analyzed under the supervision of the Rutgers University Institutional Review Board.
Detailed methods are described in supplemental Methods.
Results
MB1-47 acts as a mitochondrial uncoupler in T-ALL cells
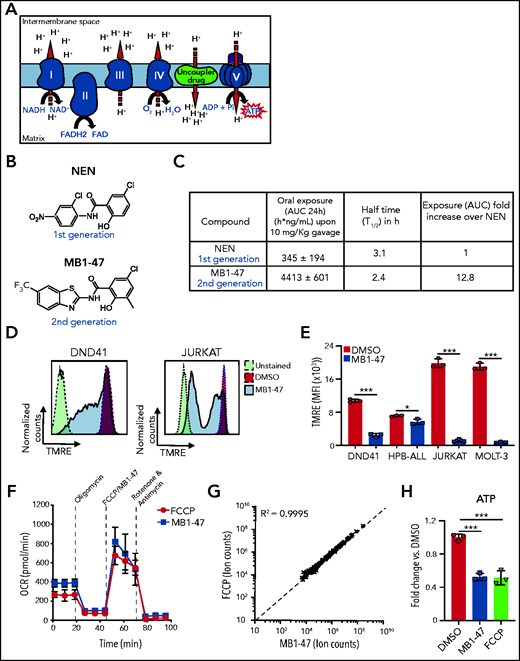
Mitochondrial metabolism is critically involved in the control of bioenergetic and biosynthetic molecular pathways to sustain tumor cell survival and proliferation.26 Specifically, previous studies showed that T-ALL cells rely on OXPHOS to maintain their proliferative capability.25 OXPHOS is coupled to protein complexes of the electron transport chain (ETC) to transfer electrons from reducing equivalents 1,4-dihydronicotinamide adenine dinucleotide (NADH) and dihydroflavin-adenine dinucleotide (FADH2) to oxygen, the final electron acceptor. As a consequence of this electron flux, the ETC generates a high proton gradient across the mitochondrial inner membrane that is required to drive adenosine triphosphate (ATP) synthesis (Figure 1A). Uncoupling drugs reduce this proton gradient and compromise the energy efficiency of mitochondria, leading to a futile oxidation of acetyl coenzyme A (acetyl-CoA) without generating ATP (Figure 1A). Niclosamide (5-chloro-salicyl-[2-chloro-4-nitro] anilide) is an oral US Food and Drug Administration (FDA)-approved drug for anthelmintic treatment with mitochondrial-uncoupling properties. Several studies indicate that niclosamide may have broad therapeutic applications for the management of more diverse diseases, including diabetes,27 viral infections, or cancer.28 Specifically, niclosamide presents a potent antiproliferative activity in a broad spectrum of cancer cell lines in vitro, being one of the top therapeutic candidates in the NCI-60 human cancer cell line screen29; its clinical potential is being assessed in ongoing clinical trials for colon cancer.30 Notably, the action of niclosamide as a mitochondrial uncoupler is well tolerated in normal cells31,32 and, used in its ethanolamine salt form (niclosamide ethanolamine [NEN]), shows promising results in liver and metastatic colon cancer in preclinical mouse models in vivo.33,34 However, both drugs have intrinsic limitations with transient and mild activities due to limited pharmacokinetic properties. Indeed, a clinical trial in prostate cancer concluded that oral niclosamide would not be viable for the treatment of castration-resistant prostate cancer.35 To improve the translational applicability of mitochondrial uncouplers, we synthesized a series of niclosamide-related analogs, leading to the identification of MB1-47, in which 1 benzene ring is substituted by a benzothiazole group, whereas a methyl group is added to the other benzene ring (Figure 1B; supplemental Figure 1A). Notably, MB1-47 shows vastly improved pharmacokinetic properties and increases in vivo exposure of the drug ∽10-fold over NEN (Figure 1C).
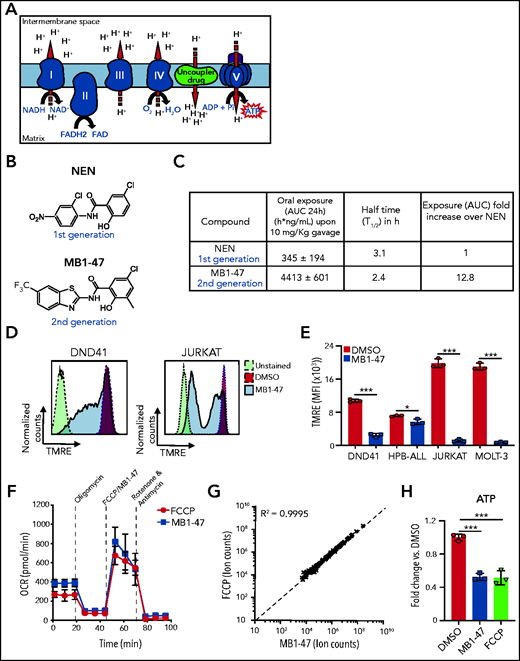
Chemical structure and mitochondrial uncoupling properties of MB1-47. (A) Schematic illustration of the ETC and the effects of uncoupling depolarizing the mitochondrial inner membrane. (B) Chemical structure of NEN and MB1-47, niclosamide-based first- and second-generation mitochondrial uncouplers, respectively. (C) Pharmacokinetic properties of NEN and MB1-47 in mice in vivo. (D) Representative flow cytometry histograms showing mitochondrial membrane potential measured by tetramethylrhodamine ethyl ester (TMRE) staining in live DND41 and JURKAT cells after 72 hours of treatment with MB1-47 (4 μM and 2 μM, respectively). (E) Geometric mean ± standard deviation (SD) quantification of TMRE staining from triplicates in 2 PTEN+ (DND41 and HPB-ALL) and 2 PTEN− (JURKAT and MOLT-3) cell lines upon 72 hours of MB-47 treatment. (F) Oxygen consumption rate (OCR) in DND41 cells, under basal conditions or in response to the indicated mitochondrial inhibitors, measured in real time using a Seahorse XF24 instrument. Data are presented as mean ± SD of n = 4 wells. (G) Metabolite levels in FCCP-treated vs MB1-47–treated DND41 cells for 24 hours (mean; n = 3). (H) Relative quantification of ATP levels from DND41 triplicates treated with DMSO, FCCP, or MB1-47 for 24 hours (mean ± SD). The goodness of fit (R2) was determined by using a simple linear regression model. Statistical significance (P) was determined by using an unpaired, 2-tailed Student t test. *P < .05; ***P < .005. AUC, area under the curve; MFI, mean fluorescence intensity.
Chemical structure and mitochondrial uncoupling properties of MB1-47. (A) Schematic illustration of the ETC and the effects of uncoupling depolarizing the mitochondrial inner membrane. (B) Chemical structure of NEN and MB1-47, niclosamide-based first- and second-generation mitochondrial uncouplers, respectively. (C) Pharmacokinetic properties of NEN and MB1-47 in mice in vivo. (D) Representative flow cytometry histograms showing mitochondrial membrane potential measured by tetramethylrhodamine ethyl ester (TMRE) staining in live DND41 and JURKAT cells after 72 hours of treatment with MB1-47 (4 μM and 2 μM, respectively). (E) Geometric mean ± standard deviation (SD) quantification of TMRE staining from triplicates in 2 PTEN+ (DND41 and HPB-ALL) and 2 PTEN− (JURKAT and MOLT-3) cell lines upon 72 hours of MB-47 treatment. (F) Oxygen consumption rate (OCR) in DND41 cells, under basal conditions or in response to the indicated mitochondrial inhibitors, measured in real time using a Seahorse XF24 instrument. Data are presented as mean ± SD of n = 4 wells. (G) Metabolite levels in FCCP-treated vs MB1-47–treated DND41 cells for 24 hours (mean; n = 3). (H) Relative quantification of ATP levels from DND41 triplicates treated with DMSO, FCCP, or MB1-47 for 24 hours (mean ± SD). The goodness of fit (R2) was determined by using a simple linear regression model. Statistical significance (P) was determined by using an unpaired, 2-tailed Student t test. *P < .05; ***P < .005. AUC, area under the curve; MFI, mean fluorescence intensity.
Next, we evaluated the uncoupling activity of MB1-47 in T-ALL cells, and we confirmed that MB1-47 treatment led to an expected and significant reduction in mitochondrial membrane potential in all cell lines tested (Figure 1D-E). In addition, similarly to carbonyl cyanide-P-trifluoromethoxyphenylhydrazone (FCCP; the gold-standard mitochondrial uncoupling drug), MB1-47 promoted an increase in oxygen consumption rate (OCR) even when ATP synthase is inhibited in presence of oligomycin (Figure 1F). Moreover, metabolomic analyses of DND41 cells treated with MB1-47 or FCCP for 24 hours revealed a very high correlation in the metabolic alterations in the presence of both drugs at their relative half-maximal inhibitory concentration (Figure 1G; supplemental Figure 1B). These changes included a significant reduction in ATP levels (Figure 1H), consistent with their uncoupling properties. Collectively, these data demonstrate that MB1-47 behaves as a mitochondrial uncoupling drug in T-ALL cells.
MB1-47 treatment blocks T-ALL cell proliferation in vitro
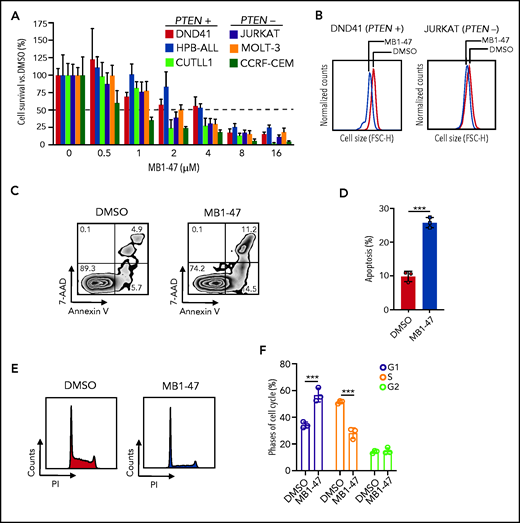
To evaluate how leukemic cells respond to mitochondrial uncoupling, we treated a panel of human T-ALL cell lines, either PTEN+ (DND41, HPB-ALL and CUTLL1)23 or PTEN− (JURKAT, MOLT-3 and CCRF-CEM)23 cells, with MB1-47 because loss of PTEN is known to confer resistance to glucocorticoids36 and to anti-NOTCH1 treatments.23 These experiments revealed that MB1-47 has potent intrinsic antileukemic activity in all cell lines tested, with a half-maximal inhibitory concentration of ∽2 to 4 μM (Figure 2A). Moreover, analysis of cellular sizes after MB1-47 treatment in vitro showed a marked reduction in cell diameters (Figure 2B; supplemental Figure 2A), a common readout of antileukemic activity.23,24 Finally, we observed similar antileukemic effects in non-NOTCH1–driven T-ALL cell lines (LOUCY and TALL-1) (supplemental Figure 2B), overall suggesting that MB1-47 could be effective in a broad set of T-ALLs. To dissect the antiproliferative effects of MB1-47, we next evaluated its impact on apoptosis and cell-cycle progression in DND41 cells. These analyses revealed a combination of cytotoxic and cytostatic effects, as MB1-47 treatment results in markedly increased apoptosis (Figure 2C-D) together with simultaneous cell-cycle arrest in G1 (Figure 2E-F). These results were confirmed in a broader panel of T-ALL cell lines, including PTEN+ and PTEN− cells, which showed less drastic G1 arrest at the expense of a block in S phase instead (supplemental Figure 2C-D), which might be reflective of the increased proliferation rate and/or hyperglycolytic phenotype in basal conditions of T-ALL cells with PTEN loss.23 Altogether, our data demonstrate that MB1-47 has strong intrinsic antileukemic activity in vitro.

MB1-47 antileukemic effects in T-ALL cell lines in vitro. (A) Relative cell survival of 6 independent human T-ALL cell lines (including PTEN+ and PTEN− cells) in the presence of MB1-47 at the indicated concentrations for 72 hours (mean ± SD; n = 3). (B) Representative flow cytometry histograms showing cell-size changes in G1-gated DND41 and JURKAT cells treated with DMSO (control) or MB1-47 (4 or 2 µM, respectively) for 72 hours. (C) Representative flow cytometry plots of annexin V (apoptotic cells) and 7-aminoactinomycin D (7-AAD; dead cells) staining. Numbers in quadrants indicate percentage of cells. (D) Quantification of apoptosis from DND41 triplicates treated for 72 hours with DMSO (control) or 4 μM MB1-47 (mean ± SD). Statistical significance (P) was determined by using the 2-tailed Student t test; ***P < .005. (E-F) Flow cytometry representation (E) and quantification (F) of cell-cycle analysis of DND41 cells treated with DMSO (control) or MB1-47 (4 μM) for 72 hours (n = 3; mean ± SD). P values in panel F were calculated using 2-way analysis of variance (ANOVA) for multiple comparisons. ***P < .005. FSC-H, forward scatter height; PI, propidium iodide.
MB1-47 antileukemic effects in T-ALL cell lines in vitro. (A) Relative cell survival of 6 independent human T-ALL cell lines (including PTEN+ and PTEN− cells) in the presence of MB1-47 at the indicated concentrations for 72 hours (mean ± SD; n = 3). (B) Representative flow cytometry histograms showing cell-size changes in G1-gated DND41 and JURKAT cells treated with DMSO (control) or MB1-47 (4 or 2 µM, respectively) for 72 hours. (C) Representative flow cytometry plots of annexin V (apoptotic cells) and 7-aminoactinomycin D (7-AAD; dead cells) staining. Numbers in quadrants indicate percentage of cells. (D) Quantification of apoptosis from DND41 triplicates treated for 72 hours with DMSO (control) or 4 μM MB1-47 (mean ± SD). Statistical significance (P) was determined by using the 2-tailed Student t test; ***P < .005. (E-F) Flow cytometry representation (E) and quantification (F) of cell-cycle analysis of DND41 cells treated with DMSO (control) or MB1-47 (4 μM) for 72 hours (n = 3; mean ± SD). P values in panel F were calculated using 2-way analysis of variance (ANOVA) for multiple comparisons. ***P < .005. FSC-H, forward scatter height; PI, propidium iodide.
MB1-47 treatment alters the metabolic landscape of T-ALL cells in vitro
Although mitochondrial uncoupling drugs perturb OXPHOS-driven ATP production, their consequences on the global metabolic landscape of cancer cells are still unclear and their potential therapeutic benefit remains controversial. We first examined the bioenergetic status of MB1-47–treated leukemic cells. Interestingly, exposure of T-ALL cells to MB1-47 for 24 hours promoted increased glucose consumption (Figure 3A) and lactate secretion (Figure 3B); this phenotype is common to all T-ALL cell lines evaluated (supplemental Figure 3A-B), indicating a metabolic rewiring toward a highly glycolytic phenotype, as previously described in metformin-treated non–small cell lung cancer cells.37 To further dissect MB1-47 metabolic impact, we next performed untargeted liquid-chromatography mass spectrometry (LC-MS) analysis of intracellular water-soluble metabolites. Exposure to MB1-47 for 24 hours caused profound alterations in the metabolic landscape of DND41 cells, leading to significant differences in 61 metabolites (Figure 3C). Importantly, even if uncoupler drugs do not directly inhibit any component of the electronic transport chain, MB1-47 decreased cellular NAD+/NADH and pyruvate/lactate ratios (Figure 3D; supplemental Figure 3C), suggesting an electron acceptor insufficiency similar to the metabolic perturbations caused by ETC inhibitors.38,39 However, MB1-47–treated-T-ALL cells were unable to fully compensate this NAD+/NADH imbalance despite the increased lactate production (Figure 3B). We next investigated the impact of MB1-47 on oxidative metabolism pathways. We observed that tricarboxylic acid (TCA) intermediates were among the most significantly altered metabolites (supplemental Figure 3D). Specifically, the levels of aconitate, citrate, α-ketoglutarate, glutamate, malate, fumarate, and acetyl-CoA drop substantially upon MB1-47 exposure (Figure 3E). Unexpectedly, we also observed a slight but significant accumulation in the intracellular levels of succinate, similar to what has been described in hypoxic ischemic tissues in vivo.40 In addition, we detected significant changes in several amino acids (Figure 3F; supplemental Figure 3D), with aspartate dropping ∽2.5-fold after MB1-47 treatment (Figure 3G). Because aspartate deficiency should impair de novo synthesis of nucleotides, we next quantified nucleotide pools and observed a significant reduction in the cellular levels of ATP, cytidine triphosphate (CTP), guanosine triphosphate [GTP], and uridine triphosphate (UTP) and an accumulation of their metabolic precursors (AMP, inosine monophosphate [IMP], cytidine monophosphate [CMP], guanosine monophosphate [GMP], and uridine monophosphate [UMP]) (Figure 3H; supplemental Figure 3D). Consistent with the increased UMP/UTP ratio (Figure 3H), and given that UTP is required for the synthesis of uridine diphosphate (UDP) sugars, we observed a significant depletion of UDP-glucose and UDP–N-acetyl-glucosamine (Figure 3I), suggesting that the hexosamine biosynthetic pathway could also be required to support T-ALL cell growth. Most of these perturbations in nucleotide biosynthesis were common to a larger panel of T-ALL cell lines upon MB1-47 treatment (supplemental Figure 3E-G). Taken together, our data show that nucleotide synthesis is severely compromised in the presence of MB1-47.
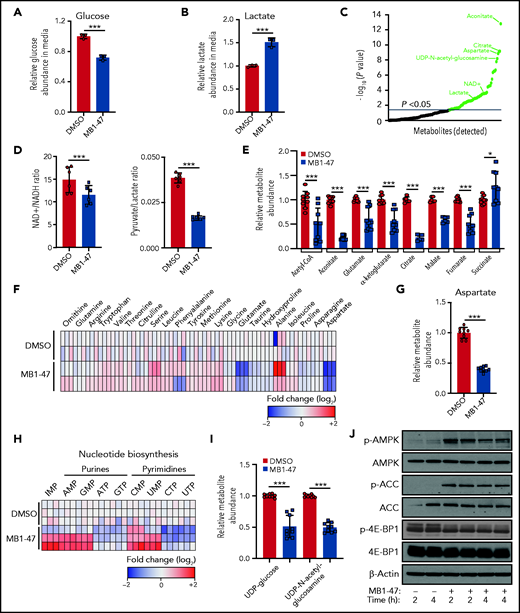
MB1-47 depletes TCA intermediates and NTPs in T-ALL cells. (A) Relative glucose abundance in media from DND41 cells cultured in the presence or absence of MB1-47 (mean ± SD; n = 3). (B) Relative lactate abundance in media from DND41 cells cultured in the presence or absence of MB1-47 (mean ± SD; n = 3). (C) Significantly altered metabolites after MB1-47 exposure, ranked by P value (−log10 transformed). (D) Intracellular ratio of NAD+/NADH (left) and pyruvate/lactate (right) in DND41 cells cultured in the presence or absence of MB1-47 (n = 6, from 2 independent replicates; bar graphs represent mean ± SD). (E) Relative abundance of indicated TCA intermediates in DND41 cells cultured in the presence or absence of MB1-47 (n = 9, from 3 independent experiments). (F) Heat map showing differential intracellular amino acid abundances (log2) after MB-47 treatment, relative to DMSO-treated (control) cells. (G) Relative abundance of aspartate in DND41 cells cultured in the presence or absence of MB1-47 (n = 9, from 3 independent experiments; mean ± SD). (H) Heat map showing differential intracellular nucleotide abundance (log2) after exposure to MB1-47, relative to DMSO-treated (control) cells. (I) Relative abundance of indicated UDP sugars in DND41 cells cultured in the presence or absence of MB1-47. All measurements were determined after MB1-47 (4 μM) treatment of 24 hours and are relative to DMSO-treated (control) cells. (J) Immunoblot analyses of AMPK, ACC, and 4E-BP1 in DND41 cells treated with DMSO or MB1-47 (4 μM) for either 2 or 4 hours. Statistical significance (P) was determined by using the 2-tailed Student t test. *P < .05; ***P < .005.
MB1-47 depletes TCA intermediates and NTPs in T-ALL cells. (A) Relative glucose abundance in media from DND41 cells cultured in the presence or absence of MB1-47 (mean ± SD; n = 3). (B) Relative lactate abundance in media from DND41 cells cultured in the presence or absence of MB1-47 (mean ± SD; n = 3). (C) Significantly altered metabolites after MB1-47 exposure, ranked by P value (−log10 transformed). (D) Intracellular ratio of NAD+/NADH (left) and pyruvate/lactate (right) in DND41 cells cultured in the presence or absence of MB1-47 (n = 6, from 2 independent replicates; bar graphs represent mean ± SD). (E) Relative abundance of indicated TCA intermediates in DND41 cells cultured in the presence or absence of MB1-47 (n = 9, from 3 independent experiments). (F) Heat map showing differential intracellular amino acid abundances (log2) after MB-47 treatment, relative to DMSO-treated (control) cells. (G) Relative abundance of aspartate in DND41 cells cultured in the presence or absence of MB1-47 (n = 9, from 3 independent experiments; mean ± SD). (H) Heat map showing differential intracellular nucleotide abundance (log2) after exposure to MB1-47, relative to DMSO-treated (control) cells. (I) Relative abundance of indicated UDP sugars in DND41 cells cultured in the presence or absence of MB1-47. All measurements were determined after MB1-47 (4 μM) treatment of 24 hours and are relative to DMSO-treated (control) cells. (J) Immunoblot analyses of AMPK, ACC, and 4E-BP1 in DND41 cells treated with DMSO or MB1-47 (4 μM) for either 2 or 4 hours. Statistical significance (P) was determined by using the 2-tailed Student t test. *P < .05; ***P < .005.
To dissect the molecular mechanism behind this metabolic stress, we evaluated the status of AMPK in DND41 cells after short treatment with MB1-47. AMPK is activated under conditions of energy stress such as nutrient deprivation or hypoxia and, through the inhibition of mammalian target of rapamycin (mTOR), acts a metabolic checkpoint suppressing anabolic biosynthetic processes.41 Our results show that MB1-47 promotes increased phosphorylation of AMPKα with concomitant phosphorylation of its downstream effector acetyl-CoA carboxylase (ACC) (Figure 3J; supplemental Figure 4A). In line with this, we also observed lower levels of phosphorylated and total 4E-BP1, supporting decreased mTOR activity (Figure 3J; supplemental Figure 4A). In addition, similar results were obtained in 2 additional T-ALL cell lines (supplemental Figure 4B), ensuring a common molecular mechanism driven by MB1-47 exposure. Finally, to investigate other potential mechanisms mediating MB1-47 effects, we also performed gene-expression profiling (RNA sequencing [RNA-seq]) of DND41 cells treated with dimethyl sulfoxide (DMSO) or MB1-47 for 24 hours. These analyses revealed that MB1-47 results in 635 significantly downregulated and 414 significantly upregulated genes (fold change >2 and adjusted P < .0001 cutoffs; supplemental Table 1). Functional annotation of genes downregulated by MB1-47 exposure identified a drastic repression of pathways related with OXPHOS, nucleotide metabolism, and messenger RNA (mRNA) stability, together with a concomitant downmodulation in DNA replication and cell-cycle progression pathways (supplemental Figure 4C; supplemental Table 2). Conversely, upregulated genes were involved in pathways related to endoplasmic reticulum stress, cell-cycle arrest, and activation of autophagy/cell catabolism (supplemental Figure 4D; supplemental Table 2). Moreover, we also identified increased levels in RFN152, SESN2, and SESN3 mRNAs (supplemental Figure 4D-E; supplemental Table 1), which encode for 3 well-known negative regulators of mTOR complex 1 signaling.42-44 In line with this, we observed transcriptional repression of genes involved in mRNA translation and ribosome biogenesis (supplemental Figure 4C-E; supplemental Table 1), 2 essential processes that are also responsive to mTOR complex 1 activity.45-47 These results are consistent with our metabolomic and proliferation results, and underscore overall that MB1-47–promoted metabolic stress might be sensed by AMPK, driving mTOR downregulation, hampering anabolic pathways, and restricting cell-cycle progression.
MB1-47 increases glucose flux and limits anaplerotic contributions to the TCA cycle
To further understand MB1-47–driven effects on the TCA cycle, untreated or MB1-47–treated cells were cultured in medium containing uniformly labeled [U-13C] glucose (Figure 4A) or [U-13C] glutamine (Figure 4B), and 13C labeling of TCA intermediates was analyzed by LC-MS. Our data indicate that T-ALL cells increased the relative glycolytic flux to TCA in the presence of MB1-47 (Figure 4C). Interestingly, examination of 13C isotopomer distribution after 4 hours of labeling revealed that MB1-47 increased the fractional enrichments of m+4 aconitate and m+3 α-ketoglutarate (Figure 4C), which can only be synthesized after a second round of the TCA cycle, suggesting an accelerated TCA turnover. Consistently, [U-13C] glutamine tracing experiments revealed an increment in the fractional enrichments of m+3 glutamate, m+3 α-ketoglutarate, and m+2 malate (Figure 4D). In addition, m+5 aconitate and m+3 malate (Figure 4D) are reduced, suggesting that isocitrate dehydrogenase has decreased reverse flux and that TCA cycle is more forward-driven upon MB1-47 treatment. Moreover, MB1-47 reduced the flux through the glutamate dehydrogenase relative to the flux through pyruvate dehydrogenase (76.56 in control cells vs 62.92 in MB1-47–treated cells) (Figure 4E), overall indicating a reduced contribution of glutamine into the TCA cycle. Finally, consistent with the increased glycolytic flux (Figure 4C), these metabolic flux analyses upon MB1-47 treatment revealed increased citrate synthase activity (Figure 4E) together with reduced pyruvate carboxylase (PC) and ATP-citrate lyase (ACLY) fluxes, and both PC and ACLY are ATP-dependent rate-limiting enzymes48,49 (Figure 4E). Collectively, our data suggest that mitochondrial ATP depletion upon MB1-47 treatment may result in increased glycolytic flux and reduced contribution of glutamine to the TCA cycle which, conversely, shows an accelerated turnover.
![MB1-47 promotes increased glycolysis and glucose flux into the TCA cycle. (A) Schematic diagram of TCA cycle intermediates 13C-labeling patterns in cells cultured with [U-13C]glucose. (B) Schematic diagram of TCA cycle intermediates 13C-labeling patterns in cells cultured with [U-13C]glutamine. (C) Isotopologs for the indicated metabolites after 4 hours of [U-13C]glucose labeling in DND41 triplicates treated with MB1-47 or DMSO (control). (D) Isotopologs for the indicated metabolites after 4 hours of [U-13C]glutamine labeling in DND41 triplicates treated with MB1-47 or DMSO (control). (E) Relative flux activity of TCA cycle in untreated (DMSO) or MB1-47–treated DND41 cells. Statistical significance (P) was determined by using multiple Student t tests. #P < .1; *P < .05; **P < .01; ***P < .005. ac-CoA, acetyl coenzyme A; CS, citrate synthase; FH, fumarate hydratase; fwd, forward; GDH, glutamate dehydrogenase; GLS, glutaminase; GOT, glutamic oxaloacetic transaminase; IDH, isocitrate dehydrogenase; α-KG, α-ketoglutarate; MDH, malate dehydrogenase; OGDH, oxoglutarate dehydrogenase; PC, pyruvate carboxylase; PDH, pyruvate dehydrogenase; rv, reverse; SDH, succinate dehydrogenase.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/138/15/10.1182_blood.2020008955/6/m_bloodbld2020008955f4.png?Expires=1766247669&Signature=0UHDAUnGue5J9Ed~P7DMqlvqH7lLOyi8P0~vcM4sTXsA1nFgq0GJalTYtoBejmsDSsoeUYd1KL4bizHiWrHzzefaaHca8UE-trIXAj6~A0HMYGYc05hq1VZm2jIKAAQ9OfXX5dawfdaTBC2Df1OSEQ6mr1gajyltNhI2iplCKhNFt1W0IPT9ZJMwexG3v66pK~SEL~g6cLBnXCl1Vx~w3nR9W3rEAEWUvp8GPNhfQH4sj54TQMrW6o4S1EqMD039LYwkjrgBu8ryXn72xBhkoEEQGeUu22vDaZ5aeLLSdvuCa64CBW0zP5eNPu2EpEU8CGbPuzg7Vj6ZRgH1YG0H3A__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
MB1-47 promotes increased glycolysis and glucose flux into the TCA cycle. (A) Schematic diagram of TCA cycle intermediates 13C-labeling patterns in cells cultured with [U-13C]glucose. (B) Schematic diagram of TCA cycle intermediates 13C-labeling patterns in cells cultured with [U-13C]glutamine. (C) Isotopologs for the indicated metabolites after 4 hours of [U-13C]glucose labeling in DND41 triplicates treated with MB1-47 or DMSO (control). (D) Isotopologs for the indicated metabolites after 4 hours of [U-13C]glutamine labeling in DND41 triplicates treated with MB1-47 or DMSO (control). (E) Relative flux activity of TCA cycle in untreated (DMSO) or MB1-47–treated DND41 cells. Statistical significance (P) was determined by using multiple Student t tests. #P < .1; *P < .05; **P < .01; ***P < .005. ac-CoA, acetyl coenzyme A; CS, citrate synthase; FH, fumarate hydratase; fwd, forward; GDH, glutamate dehydrogenase; GLS, glutaminase; GOT, glutamic oxaloacetic transaminase; IDH, isocitrate dehydrogenase; α-KG, α-ketoglutarate; MDH, malate dehydrogenase; OGDH, oxoglutarate dehydrogenase; PC, pyruvate carboxylase; PDH, pyruvate dehydrogenase; rv, reverse; SDH, succinate dehydrogenase.
MB1-47 promotes increased glycolysis and glucose flux into the TCA cycle. (A) Schematic diagram of TCA cycle intermediates 13C-labeling patterns in cells cultured with [U-13C]glucose. (B) Schematic diagram of TCA cycle intermediates 13C-labeling patterns in cells cultured with [U-13C]glutamine. (C) Isotopologs for the indicated metabolites after 4 hours of [U-13C]glucose labeling in DND41 triplicates treated with MB1-47 or DMSO (control). (D) Isotopologs for the indicated metabolites after 4 hours of [U-13C]glutamine labeling in DND41 triplicates treated with MB1-47 or DMSO (control). (E) Relative flux activity of TCA cycle in untreated (DMSO) or MB1-47–treated DND41 cells. Statistical significance (P) was determined by using multiple Student t tests. #P < .1; *P < .05; **P < .01; ***P < .005. ac-CoA, acetyl coenzyme A; CS, citrate synthase; FH, fumarate hydratase; fwd, forward; GDH, glutamate dehydrogenase; GLS, glutaminase; GOT, glutamic oxaloacetic transaminase; IDH, isocitrate dehydrogenase; α-KG, α-ketoglutarate; MDH, malate dehydrogenase; OGDH, oxoglutarate dehydrogenase; PC, pyruvate carboxylase; PDH, pyruvate dehydrogenase; rv, reverse; SDH, succinate dehydrogenase.
MB1-47 treatment is well tolerated in vivo
To test the potential relevance of our findings in vivo, we first checked MB1-47 effects in healthy mice fed with a control diet or a diet containing MB1-47 for >30 consecutive days. Importantly, MB1-47 treatment is well tolerated, as revealed by the unchanged mouse weight during the entire experiment (supplemental Figure 5A). Moreover, mice did not present any sign of anemia or other hematologic alterations (supplemental Figure 5B). Furthermore, detailed immunophenotypic analyses of T-cell development in these mice revealed no significant differences in the percentages of different thymocyte populations (supplemental Figure 5C-F), including CD4/CD8 double-negative (DN1, DN2, DN3, and DN4) (supplemental Figure 5E-F), CD4/CD8 double-positive (supplemental Figure 5C-D), or single-positive CD4 or CD8 cells (supplemental Figure 5C-D). Therefore, our data suggest that there might be a good therapeutic window for the use of MB1-47 in vivo.
Acute MB1-47 treatment altered the metabolic profile of T-ALL in vivo
Next, we investigated the metabolic impact of MB1-47 in T-ALL in vivo, taking advantage of our previously established NOTCH1-induced mouse primary leukemias.24 Mice showing signs of overt leukemia (>60% green fluorescent protein–positive [GFP+] leukemic cells in peripheral blood) were exposed to MB1-47 or vehicle as a control over a 4-hour period (Figure 5A and “Methods”), followed by untargeted metabolomic analyses (LC-MS) of leukemic spleens. Even after this short time of exposure, we detected significant differences in 20 metabolites between untreated and MB1-47–treated leukemias (Figure 5B). Remarkably, ∽60% of detectable metabolites were also significantly altered after MB1-47 treatment in vitro, indicating that MB1-47 promotes a similar metabolic impact in vivo. Indeed, we also observed a slight reduction in the pyruvate/lactate ratio (Figure 5C), despite the potential interference from nonleukemic stromal cells. Remarkably, and consistent with our previous in vitro data, we detected a significant reduction in the levels of aspartate, glutamate, and malate (Figure 5D), indicating that these metabolic perturbations constitute the core of the immediate response to uncoupler drugs and may mediate the first steps in the metabolic cascade for its antiproliferative activity. Indeed, acute MB1-47 treatment also led to cytotoxic effects in T-ALL in vivo, as shown by a marked increase in poly (ADP-ribose) polymerase (PARP) cleavage (Figure 5E; supplemental Figure 6A). Mechanistically, MB1-47 treatment in primary leukemias in vivo resulted in increased AMPKα phosphorylation, together with a concomitant phosphorylation of its effector ACC and reduced levels of phosphorylated 4E-BP1, indicating a downregulation of mTOR activity (Figure 5F; supplemental Figure 6B). In line with this, MB1-47 treatment for 3 consecutive days (Figure 6A) effectively blocked leukemic cell growth and reduced tumor burden, as revealed by decreased spleen weight and significantly reduced number of GFP+ leukemic cells both in spleen and bone marrow (Figure 6B-D). In addition, our analyses confirmed a combination of cytotoxic and cytostatic effects in these leukemias (Figure 6E-F; supplemental Figure 6C), consistent with our previous results with MB1-47 in T-ALL cell lines in vitro.

Metabolic and signaling effects upon acute MB1-47 treatment of leukemic mice in vivo. (A) Schematic illustration of acute exposure to MB1-47 in vivo via gavage (detailed in “Methods”). (B) Significantly altered metabolites after acute exposure to MB1-47, ranked by P value (−log2 transformed). (C) Ratio of pyruvate/lactate abundances from leukemic spleens after acute treatment with MB1-47 in vivo (mean ± SD). (D) Relative abundance of the indicated metabolites in leukemic spleens after acute treatment with MB1-47 in vivo. (E) Immunoblot analyses of cleaved PARP in leukemic spleens after acute treatment with MB1-47 in vivo. (F) Immunoblot analyses of AMPK, ACC, and 4E-BP1 in leukemic spleens after acute treatment with MB1-47 in vivo. Statistical significance (P) was determined by using the 2-tailed Student t test. **P < .01; ***P < .005.
Metabolic and signaling effects upon acute MB1-47 treatment of leukemic mice in vivo. (A) Schematic illustration of acute exposure to MB1-47 in vivo via gavage (detailed in “Methods”). (B) Significantly altered metabolites after acute exposure to MB1-47, ranked by P value (−log2 transformed). (C) Ratio of pyruvate/lactate abundances from leukemic spleens after acute treatment with MB1-47 in vivo (mean ± SD). (D) Relative abundance of the indicated metabolites in leukemic spleens after acute treatment with MB1-47 in vivo. (E) Immunoblot analyses of cleaved PARP in leukemic spleens after acute treatment with MB1-47 in vivo. (F) Immunoblot analyses of AMPK, ACC, and 4E-BP1 in leukemic spleens after acute treatment with MB1-47 in vivo. Statistical significance (P) was determined by using the 2-tailed Student t test. **P < .01; ***P < .005.

MB1-47 shows AMPK-mediated antileukemic effects in mouse primary leukemias in vivo. (A) Schematic illustration of treatment with MB1-47–containing diet in leukemic mice in vivo for 3 consecutive days (detailed in “Methods”; n = 5). (B-D) Changes in leukemia burden after 3 days of MB1-47 treatment, as assessed by total spleen weight (B) or by flow cytometry detection of leukemic GFP+ cells in spleen (C) and bone marrow (D). Statistical significance (P) was determined by using the 1-tailed Student t test. *P < .05; ***P < .005. (E) Immunoblot analyses of cleaved PARP in leukemic spleens after 3 days of MB1-47 treatment. (F) Flow cytometry representation of cell-cycle analysis of T-ALL cells from leukemic spleens after 3 days of MB1-47 treatment. P values were calculated using 2-way ANOVA for multiple comparisons; ***P < .005. (G-H) Kaplan-Meier survival curves of mice harboring isogenic Pten+ (G) and Pten− (H) T-ALLs treated with an MB1-47–containing diet or a control diet. (I) Schematic illustration of bone marrow progenitor-retroviral transduction protocol for the generation and analysis of NOTCH1-induced Ampk-conditional knockout T-ALLs. (J) Kaplan-Meier survival curves of mice harboring Ampk+ and Ampk− isogenic leukemias treated with an MB1-47–containing diet or a control diet. P values in survival curves were calculated with the log-rank test; n = 10 mice per group. ***P < .005.
MB1-47 shows AMPK-mediated antileukemic effects in mouse primary leukemias in vivo. (A) Schematic illustration of treatment with MB1-47–containing diet in leukemic mice in vivo for 3 consecutive days (detailed in “Methods”; n = 5). (B-D) Changes in leukemia burden after 3 days of MB1-47 treatment, as assessed by total spleen weight (B) or by flow cytometry detection of leukemic GFP+ cells in spleen (C) and bone marrow (D). Statistical significance (P) was determined by using the 1-tailed Student t test. *P < .05; ***P < .005. (E) Immunoblot analyses of cleaved PARP in leukemic spleens after 3 days of MB1-47 treatment. (F) Flow cytometry representation of cell-cycle analysis of T-ALL cells from leukemic spleens after 3 days of MB1-47 treatment. P values were calculated using 2-way ANOVA for multiple comparisons; ***P < .005. (G-H) Kaplan-Meier survival curves of mice harboring isogenic Pten+ (G) and Pten− (H) T-ALLs treated with an MB1-47–containing diet or a control diet. (I) Schematic illustration of bone marrow progenitor-retroviral transduction protocol for the generation and analysis of NOTCH1-induced Ampk-conditional knockout T-ALLs. (J) Kaplan-Meier survival curves of mice harboring Ampk+ and Ampk− isogenic leukemias treated with an MB1-47–containing diet or a control diet. P values in survival curves were calculated with the log-rank test; n = 10 mice per group. ***P < .005.
MB1-47 significantly extends survival in mouse T-ALL via activation of AMPK in vivo
Because MB1-47 showed a favorable safety profile together with promising antileukemic effects, we next examined whether MB1-47 could extend the survival of mice harboring NOTCH1-induced primary leukemias in vivo. Here, we took advantage of our previously established Pten-conditional knockout (Ptenflox/flox-Rosa26Cre-ERT2/+) isogenic NOTCH1-induced leukemias24 in order to analyze the effects of MB1-47 on the survival of mice harboring either Pten+ or Pten− (upon tamoxifen-induced activation of the Cre recombinase) leukemias (supplemental Figure 6D). Remarkably, MB1-47 treatment translated into very drastic antileukemic effects in vivo with significantly extended survival of mice harboring Pten+ leukemias (Figure 6G; median survival in control mice = 14 days, median survival in MB1-47 treated mice = 28 days). In addition, and most notably, these antileukemic effects were also maintained in mice harboring Pten− leukemias (Figure 6H). Despite the expected acceleration of disease kinetics upon Pten loss24 (median survival in control mice = 11 days; Figure 6H), MB1-47 treatment still conferred a similar approximately twofold increase in disease latency in mice harboring Pten-null leukemias (median survival in MB1-47–treated mice = 21 days; Figure 6H). These findings might be clinically relevant, as loss of PTEN is known to mediate resistance to glucocorticoid treatment,36 as well as resistance to anti-NOTCH1 therapies.23,24
Because our previous results demonstrated that MB1-47 treatment leads to AMPK activation, we next decided to genetically test the role of AMPK in T-ALL in vivo. To this end, we developed an isogenic model of NOTCH1-induced Ampk-conditional knockout T-ALL (Ampkflox/flox-Rosa26Cre-ERT2/+) following a similar strategy (Figure 6I). We then transplanted these Ampk-conditional knockout leukemias into secondary mice that were treated with vehicle or tamoxifen to induce isogenic loss of Ampk (supplemental Figure 6E); mice were further subdivided into animals fed with a control diet or an MB1-47–containing diet to dissect the role of AMPK in normal T-ALL progression, as well as its potential contribution to mediate the therapeutic effects observed upon MB1-47 treatment. Interestingly, our results indicate that AMPK is dispensable for normal leukemic progression, as Ampk− leukemias show the same disease kinetics as Ampk+ isogenic leukemias (Figure 6J). In this setting, MB1-47 treatment led to a marked extension in survival of mice harboring Ampk+ leukemias. However, and most notably, isogenic loss of Ampk significantly impaired the antileukemic effects of MB1-47 in vivo (Figure 6J). Overall, our results demonstrate strong antileukemic effects for MB1-47 treatment in vivo leading to significant survival extension, and underscore a major mechanistic role for AMPK in mediating mitochondrial uncoupling effects.
MB1-47 significantly extends survival in human T-ALL PDXs in vivo
Finally, to further explore the clinical relevance of mitochondrial uncoupling in T-ALL, we also tested the therapeutic efficacy of MB1-47 in human T-ALL PDXs. In this context, MB1-47 treatment in mice bearing a previously described PTEN+ and NOTCH1-mutated PDX from the TLX3 genetic subgroup (PDTALL19; supplemental Figure 7A-B)24,50,51 translated into a significant increase in survival (Figure 7A). We previously described that the therapeutic effects of MB1-47 are maintained in NOTCH1-driven Pten− mouse leukemias (Figure 6H), however, most human T-ALL cases with loss of PTEN are wild-type for NOTCH1.52 Thus, we also tested the antileukemic effects of MB1-47 in a different PDX sample that is PTEN− but wild-type for NOTCH1 and belongs to the high-risk early T-cell precursor genetic subgroup (CU-L-157; supplemental Figure 7A-B). Importantly, treatment of CU-L-157–bearing mice with MB1-47 resulted in decreased tumor burden (supplemental Figure 7C), which again translated into a significant extension in survival (Figure 7B). Finally, a pilot experiment using yet another PTEN− PDX belonging to the TAL-LMO subgroup (PDTALL9; supplemental Figure 7A-B)50,51 resulted in significantly reduced tumor burden after 1 month of MB1-47 treatment, as shown by reduced spleen weight and decreased infiltration of human CD45+ cells in both spleen and bone marrow (supplemental Figure 7D). Overall, our results demonstrate that MB1-47 could have significant antileukemic effects in human T-ALL in vivo, regardless of molecular subgroup and/or mutational status of NOTCH1 or PTEN.
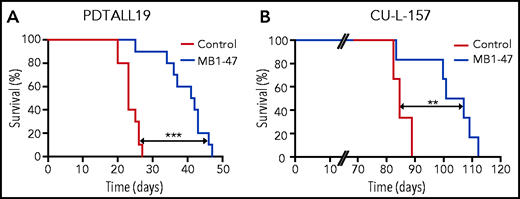
MB1-47 shows antileukemic effects in human T-ALL PDXs in vivo. (A) Kaplan-Meier survival curve of mice harboring PDTALL19, a NOTCH1-mutated PTEN+ human T- ALL PDX, treated with a control diet or an MB1-47–containing diet (n = 10 mice per group). (B) Kaplan-Meier survival curve of mice harboring CU-L-157, a NOTCH1–wild-type PTEN− human T- ALL PDX, treated with a control diet or an MB1-47–containing diet (n = 6 mice per group). P values were calculated with the log-rank test. **P < .01; ***P < .005.
MB1-47 shows antileukemic effects in human T-ALL PDXs in vivo. (A) Kaplan-Meier survival curve of mice harboring PDTALL19, a NOTCH1-mutated PTEN+ human T- ALL PDX, treated with a control diet or an MB1-47–containing diet (n = 10 mice per group). (B) Kaplan-Meier survival curve of mice harboring CU-L-157, a NOTCH1–wild-type PTEN− human T- ALL PDX, treated with a control diet or an MB1-47–containing diet (n = 6 mice per group). P values were calculated with the log-rank test. **P < .01; ***P < .005.
Discussion
The development of new therapeutic agents for the treatment of T-ALL remains an unmet clinical need. Recent advances have improved our understanding of the basic biology and genetics driving this disease.53 However, clinical management of T-ALL cases is still based on intensive salvage chemotherapy regimens. Even if these treatments have considerably improved clinical outcomes,3 many patients still relapse and/or present long-term adverse health impairments, highlighting the urgent need to discover more effective interventions with less overlapping toxicities. Oncogenic transformation, driven by MYC, RAS, NOTCH, or loss of PTEN, is closely linked to leukemia-associated metabolic rewiring and these oncogene-driven metabolic dependencies may offer a more druggable opportunity for translational purposes.10 For decades, the cancer metabolism field focused efforts on perturbing the enhanced glycolysis (Warburg effect) observed in cancer cells, however, recent studies demonstrated that mitochondrial metabolism is essential for tumorigenesis.54-57
In the present study, we developed and characterized MB1-47, a novel niclosamide-based second-generation mitochondrial-uncoupling drug, and we provide evidence that T-ALL cells are highly dependent on OXPHOS and mitochondrial ATP for proliferation and survival. Global metabolic profiling analyses revealed that MB1-47 treatment generates an environment of energy deficiency and macromolecule depletion that severely compromises nucleotide biosynthesis, leading to apoptosis and cell-cycle arrest, comparable to what has been observed after the inhibition of complex I in acute myeloid leukemia.58 Even though MB1-47 increases oxygen consumption, we observed a low NAD+/NADH ratio, similar to the metabolic phenotype in proliferating cells upon treatment with ETC inhibitors, which show an electron acceptor insufficiency.38,39 Given the mechanism of action of MB1-47, which is to promote futile mitochondrial respiration, it is reasonable to speculate that this glycolytic phenotype might be a mechanism to compensate the decrease in mitochondrial ATP levels and/or a relative decrease in electron acceptors. Beyond this NAD+ deficiency, we found that MB1-47 also reduces the levels of most TCA intermediates except succinate. In addition, MB1-47 led to significant changes in several amino acids and, consistent with the lower NAD+/NADH ratio observed, aspartate synthesis seems to be limited.38,39,59 Moreover, we observed a significant reduction of nucleotide triphosphates (NTPs), with a concomitant accumulation of nucleotide monophosphates. Thus, our data indicate that neither the aspartate deficiency nor the relative electron acceptor insufficiency caused the inhibition of nucleotide biosynthesis, as they are required for the conversion of IMP to AMP and GMP, respectively. However, subsequent reactions for the final production of NTPs are catalyzed by ATP-dependent kinases. Thus, mitochondrial ATP could be the limiting metabolite for normal cell-cycle progression in T-ALL cells treated with MB1-47. Related to this, a recent study demonstrated that mitochondrial ATP, but not glycolytic ATP, regulates fatty acid uptake and transport.60
In addition, metabolic isotope-labeling analyses indicated increased glycolytic flux and reduced contribution of glutamine to the TCA cycle which, conversely, shows an accelerated turnover. Moreover, the depletion in mitochondrial ATP led to reduced metabolic fluxes through PC and ACLY, 2 ATP-dependent rate-limiting enzymes. Indeed, in normal conditions, cells use pyruvate carboxylation to produce aspartate, and the decreased flux through PC may explain in part the reduced levels for this amino acid. Furthermore, PC deficiency also results in depletion of TCA cycle intermediates and acetyl-CoA,61 2 phenotypes induced by MB1-47.
We also demonstrate that MB1-47 is selectively toxic against leukemic cells without affecting T-cell development in the thymus or other hematologic populations in peripheral blood from healthy mice; thus, MB1-47 shows an acceptable safety profile. In addition, acute exposure of leukemic mice to MB1-47 led to depletion of some TCA intermediates, such as malate and aspartate, suggesting that the metabolic shutdown observed in vitro might be reliably translated in vivo. Importantly, MB1-47 exhibited strong antileukemic activity as a single agent and significantly extended the survival of mice harboring Pten+ leukemias. Strikingly, and unlike glutaminase inhibition,24 MB1-47 as single agent also delayed T-ALL progression in mice harboring Pten-null leukemias. Our results uncover a mitochondrial respiration dependency for Pten-null leukemias, despite the fact that PTEN loss induces a hyperglycolytic phenotype that drives resistance to NOTCH inhibitors.23,24 We also formally tested the role of AMPK in leukemia progression and response to MB1-47 by generating Ampk-conditional knockout leukemias. Notably, our results demonstrate that AMPK is dispensable for normal leukemic progression, but AMPK loss significantly impairs the therapeutic response to MB1-47 treatment. Previous studies offered conflicting reports for AMPK in leukemia. Specifically, Kishton et al suggested an oncogenic role for AMPK in T-ALL, as secondary loss of AMPK extended survival in a T-ALL model driven by intracellular activated NOTCH1 (intracellular domain of Notch1 [ICN1]).25 Conversely, a recent report suggested that AMPK could have a cell-autonomous tumor-suppressor role mediating the therapeutic effect of the complex I inhibitor phenformin (not metformin) in leukemias driven by lymphocyte-specific deletion of Pten.62 In this setting, our results might help to reconcile the different roles suggested for AMPK in T-ALL. In our study, we used a ΔE-NOTCH1 allele, which is an oncogenic form that lacks the extracellular portion of NOTCH1 but is still tethered to the membrane and requires processing by the γ-secretase complex. Thus, this model bears a closer resemblance to the mutations in NOTCH1 observed in patients with T-ALL, which typically cluster in the heterodimerization domain and in the PEST (proline, glutamic acid, serine, and threonine) domain, but still need γ-secretase processing for the release of ICN1.53 In this context, it might be possible that, using a model of supraphysiological NOTCH1 activation such as the ICN1 model, AMPK might be needed for leukemia cells to adapt and survive the strong metabolic stress imposed by the constant presence of ICN1. However, our results demonstrate that AMPK does not play a significant role in normal leukemia progression when using a weaker form of NOTCH1 that still requires γ-secretase cleavage. On the other hand, our results reveal that AMPK plays a major role in mediating the therapeutic effects of mitochondrial uncoupling drugs. Moreover, even if AMPK loss significantly impairs the therapeutic effects of MB1-47, it is worth noting that Ampk-null leukemia-bearing mice treated with MB1-47 still show a reduced, but significant, extension in survival as compared with control mice. This result might be due to the only partial Ampk deletion observed upon tamoxifen treatment (supplemental Figure 6E). Alternatively, it suggests that part of the therapeutic effects of MB1-47 might still be due to off-target effects. However, yet another more intriguing possibility to explain these results is that mitochondrial uncoupling might have non–cell-autonomous antileukemic effects, as leukemia-bearing recipient mice are otherwise wild-type mice that retain AMPK expression. Indeed, a very recent study demonstrated that tumor-associated myeloid cells provide critical support for T-ALL in vivo,63 thus, MB1-47–driven mitochondrial uncoupling in the myeloid compartment (or in other cells or tissues) might be partly responsible for the therapeutic effects observed. Further studies to dissect the potential non–cell-autonomous antileukemic role of mitochondrial uncoupling are therefore warranted.
Overall, our data support strong therapeutic effects for MB1-47 treatment in T-ALL. However, its therapeutic effects might well go beyond T-cell leukemia, as previous analogs showed therapeutic activity in liver and colon cancer preclinical models. In addition, a very recent study demonstrated its therapeutic potential in a model of pancreatic metastasis64 and, moreover, the NCI-60 panel showed potential therapeutic effects of niclosamide in additional types of cells, including chronic myelogenous leukemia, myeloma, lymphoma, acute promyelocytic leukemia, or acute myeloid leukemia cell lines.29 Related to this, directly targeting OXPHOS with the novel ETC complex I inhibitor IACS-010759 has shown strong antileukemic effects in acute myeloid leukemia in vivo,58 overall suggesting that mitochondrial uncoupling with MB1-47 might be an attractive therapeutic option in additional malignancies.
Finally, and specifically focusing on T-ALL, our data have been obtained using multiple cell lines in vitro, NOTCH1-driven mouse primary leukemias in vivo, and independent human PDXs from different T-ALL genetic subgroups, overall suggesting that mitochondrial uncoupling with MB1-47 could be broadly effective against different T-ALL subtypes, independent of oncogenic drivers and the mutational status of NOTCH1 or PTEN. Still, a previous study suggested that ex vivo treatment with tigecycline, an antibiotic drug that inhibits mitochondrial protein translation, could confer increased malignancy and leukemia-initiating cell activity in a TAL1 T-ALL model without chromosome 6q deletion.65 Even if the mechanisms by which tigecycline and MB1-47 perturb mitochondrial homeostasis are widely different, further analyses of MB1-47 in a more exhaustive panel of human PDXs are therefore needed to address any potential unexpected deleterious effects of MB1-47 treatment in T-ALL cases with specific mutations. Lastly, it is important to note that MB1-47 treatment as single-agent delayed leukemia progression in all settings tested but failed to achieve complete leukemia remission, and all MB1-47–treated leukemias eventually relapsed. Clinical treatment of patients with T-ALL notoriously requires intensive multiagent regimens, thus, analyzing potential synergistic combinations between MB1-47 and currently used antileukemic drugs during induction, consolidation, intensification, maintenance, and/or remission phases will be important.66,67 In this setting, we performed preliminary in vitro experiments using MB1-47 in combination with the components of the vincristine, dexamethasone, l-asparaginase regimen, and we observed either additive (vincristine) or synergistic (dexamethasone, l-asparaginase) effects driven by increased cytotoxic effects (supplemental Figure 8). Although these results are very promising, more comprehensive analyses of potential synergisms and/or added toxicities upon combination of MB1-47 with other antileukemic agents in vivo are clearly required to identify the best strategy to introduce MB1-47 in T-ALL treatment regimens. Overall, our findings support a critical role for mitochondrial OXPHOS in T-ALL and demonstrate that the novel MB1-47 mitochondrial-uncoupling drug could be an attractive therapeutic strategy for the treatment of T-ALL patients.
Acknowledgments
The authors thank all members of the D.H. Laboratory for helpful discussions. The authors are grateful to Adolfo Ferrando (Columbia University, New York, NY) for constant support. The authors thank Marta Sanchez-Martín and Marissa Rashkovan in the Ferrando laboratory, for their help with the generation and characterization of the CU-L-157 PDX sample. The authors also thank everyone involved with JuanLord for their support.
Work in the laboratory of D.H. was supported by National Institutes of Health, National Cancer Institute (NIH/NCI) grant R01CA236936, Research Scholar grant RSG-19-161-01-TBE from the American Cancer Society, an American Association for Cancer Research (AACR)–Bayer Innovation and Discovery grant, the Alex’s Lemonade Stand Foundation, the Leukemia Research Foundation, the Children’s Leukemia Research Association, and the Gabrielle’s Angel Foundation for Cancer Research. V.d.S.-D. was supported by the New Jersey Commission on Cancer Research (DCHS19PPC008). In addition, Rutgers Cancer Institute of New Jersey shared resources supported, in part, by NIH/NCI Cancer Center support grant P30CA072720 were instrumental for this project and, more specifically, the Metabolomics Shared Resource (P30CA072720-5923). B.C. was supported by Mito Biopharma. S.J. was supported by NIH/NCI grant R21CA216604.
The schematic illustrations in this manuscript (Figures 5A and 6A,I) were created with BioRender.com.
Authorship
Contribution: V.d.S.-D. performed most molecular biology, cellular, and animal experiments and wrote the manuscript; O.L. assisted with in vitro experiments; M.A., S.L., and H.T. assisted with some animal experiments; E.C. processed metabolomic samples; B.C. and D.A. designed and synthesized MB1-47; A.A. did initial mitochondrial uncoupling characterization of MB1-47; A.S. performed computational RNA-seq analyses; S.M. and S.I. generated and provided the PDTALL19 and PDTALL9 xenografts; H.K. supervised all computational analyses; X.S. supervised and analyzed all the metabolomic experiments; S.J. conceived the idea, assisted with design and synthesis of MB1-47, and assisted with design of study; and D.H. conceived the idea, designed the study, supervised the research, and wrote the manuscript with V.d.S.-D.
Conflict-of-interest disclosure: B.C., D.A., H.T., and S.J. are coinventors of the patent covering MB1-47. S.J. is a cofounder of Mito Biopharma, which licensed the patent. The remaining authors declare no competing financial interests.
Correspondence: Daniel Herranz, Rutgers Cancer Institute of New Jersey, Rutgers University, 195 Little Albany St, New Brunswick, NJ 08901; e-mail: dh710@cinj.rutgers.edu; Shengkan Jin, Department of Pharmacology, Robert Wood Johnson Medical School, Rutgers University, 675 Hoes Ln West, Piscataway, NJ 08854; e-mail: victor.jin@rutgers.edu; and Victoria da Silva-Diz, Rutgers Cancer Institute of New Jersey, Rutgers University, 195 Little Albany St, New Brunswick, NJ 08901; e-mail: md1399@cinj.rutgers.edu.
The RNA-seq data reported in this article have been deposited in the Gene Expression Omnibus database (accession number GSE165405).
The authors will provide any material, data set, or protocol without restrictions upon requesting e-mail to the corresponding authors.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.