

Key Points
lncRNA LOUP coordinates with RUNX1 to induce PU.1 long-range transcription, conferring myeloid differentiation and inhibiting cell growth.
RUNX1-ETO limits chromatin accessibility at the LOUP locus, causing inhibition of LOUP and PU.1 expression in t(8;21) AML.

Abstract
The mechanism underlying cell type-specific gene induction conferred by ubiquitous transcription factors as well as disruptions caused by their chimeric derivatives in leukemia is not well understood. Here, we investigate whether RNAs coordinate with transcription factors to drive myeloid gene transcription. In an integrated genome-wide approach surveying for gene loci exhibiting concurrent RNA and DNA interactions with the broadly expressed Runt-related transcription factor 1 (RUNX1), we identified the long noncoding RNA (lncRNA) originating from the upstream regulatory element of PU.1 (LOUP). This myeloid-specific and polyadenylated lncRNA induces myeloid differentiation and inhibits cell growth, acting as a transcriptional inducer of the myeloid master regulator PU.1. Mechanistically, LOUP recruits RUNX1 to both the PU.1 enhancer and the promoter, leading to the formation of an active chromatin loop. In t(8;21) acute myeloid leukemia (AML), wherein RUNX1 is fused to ETO, the resulting oncogenic fusion protein, RUNX1-ETO, limits chromatin accessibility at the LOUP locus, causing inhibition of LOUP and PU.1 expression. These findings highlight the important role of the interplay between cell-type–specific RNAs and transcription factors, as well as their oncogenic derivatives in modulating lineage-gene activation and raise the possibility that RNA regulators of transcription factors represent alternative targets for therapeutic development.
Introduction
Lineage-control genes that dictate cellular identities are often expressed in dynamic and hierarchical patterns.1-3 Disturbance of these established normal patterns results in anomalies.4 In the blood system, the ETS-family transcription factor PU.1 (also known as Spi-1) is essential for myeloid differentiation. PU.1 is silent in most tissues and cell types but is expressed at the highest levels in myeloid cells including granulocytes and monocytes.5 Downregulation of PU.1 impairs myeloid cell differentiation, leading to acute myeloid leukemia (AML).6,7PU.1 is a major downstream transcriptional target of Runt-related transcription factor 1 (RUNX1), which is expressed in many different cell types and plays diverse biological roles in hematopoiesis, and development of neurons, hair follicles, and skin.8-12 In AML with the t(8;21) chromosomal translocation, a portion of RUNX1 containing the Runt DNA-binding domain is fused to ETO, giving rise to the oncogenic transcription factor fusion RUNX1-ETO.13,14 Previously, we have reported that RUNX1-ETO inhibits PU.1 expression15 but the mechanism underlying this transcriptional inhibition remains to be determined. In general, how broadly expressed transcription factors, such as RUNX1, modulate cell type- and gene-specific induction and how their chimeric derivatives disrupt this normal regulation in leukemia are poorly understood.
Transcription of many cell type-specific genes is induced by enhancer elements, which are located at variable distances from gene targets.16,17 For instance, PU.1 transcription is induced by the formation of a specific chromatin loop resulting from the interaction between the upstream regulatory element (URE; −17 kb in humans and −14 kb in mice) and the proximal promoter region (PrPr).18-20 Interestingly, abrogation of RUNX1-binding motifs at the URE reduces URE-PrPr interaction, resulting in decreased PU.1 expression in myeloid cells.8,15 Because RUNX1 is broadly expressed, how this transcription factor modulates chromatin structure in such a gene- and cell type-specific manner remains unclear.
With advances in whole-transcriptome sequencing over the last decade, thousands of noncoding RNAs (ncRNAs) have been unveiled.21 Arbitrarily defined as ncRNAs of at least 200 nucleotides in length, long noncoding RNAs (lncRNAs) are implicated in displaying tissue-specific expression patterns22,23 and might undergo posttranscriptional processing such as splicing and polyadenylation.24 Through interactions with DNAs, proteins, and other RNAs, lncRNAs regulate fundamental cellular processes including transcription, RNA stability, and DNA methylation.24-26 To date, only a few lncRNAs have been precisely mapped and functionally defined,23 leaving most lncRNAs poorly annotated and largely unexplored.
In this study, we identified a myeloid-specific lncRNA, termed “long noncoding RNA originating from the URE of PU.1,” or LOUP, from an integrated genome-wide approach aimed at screening for gene loci exhibiting concurrent RNA and DNA interactions with RUNX1. We demonstrated that LOUP induces PU.1 expression, conferring myeloid differentiation, and inhibiting cell growth. LOUP serves as a central hub in opposing regulation by RUNX1 and its derived oncogenic fusion, RUNX1-ETO. Our findings provide a model explaining how a lineage gene is activated in normal myeloid development and dysregulated in leukemia.
Methods
Cell lines and cell culture
U937, HL-60, K562, HEK293T, RAW 264.7, NB4, Jurkat, Kasumi-1, and THP-1 cells were obtained from the American Type Culture Collection (ATCC). U937, HL-60, NB4, Jurkat, and K562 cells were cultured in full RPMI 1640 medium (supplemented with 10% [v/v] fetal bovine serum [FBS; Cellgro] and 1% penicillin-streptomycin). Kasumi-1 cells were cultured in the same medium plus 20% (v/v) FBS. THP-1 cells were cultured in full RPMI 1640 medium supplemented with 2-mercaptoethanol to a final concentration of 0.05 mM. HEK293T and RAW 264.7 cells were cultured in Dulbecco modified Eagle medium supplemented with 10% (v/v) FBS and 1% penicillin-streptomycin. All cells were grown at 37°C in 5% (v/v) CO2 and humidified incubators.
AML patient sample collection
Bone marrow (BM) samples were obtained from newly diagnosed patients with AML at the Tor Vergata University Hospital (Rome, Italy) with informed consent. Diagnoses were performed according to the 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia.27 BM mononuclear cells were isolated by Ficoll gradient centrifugation using Lympholyte-H (Cedarlane), according to the manufacturer’s instructions.
Methods for assaying myeloid culture of primary cells; interactions of RNA, DNA, and protein with chromatin; chromatin structure; gene-expression manipulation; and bioinformatic analyses are in supplemental Methods (available on the Blood Web site).
Results
Identification of RUNX1-interacting RNAs at myeloid gene loci
We started out by performing a transcriptome-wide survey for RUNX1-interacting RNAs in the monocytic cell line THP-1 using formaldehyde RNA immunoprecipitation (RIP) sequencing (RIP-seq).28,29 RUNX1-interacting RNAs were captured by anti-RUNX1 antibody (supplemental Figure 1A-C) and sequenced by paired-end massively parallel sequencing. By annotating 10 109 high-confident RUNX1-RIP peaks to the GRCh38.p12 gene catalog,30 we identified 6035 gene loci carrying ≥1 of these peaks (supplemental Figure 1D, left). Most of the peaks were detected within transcript bodies and promoters (supplemental Figure 1E). To identify genes exhibiting concurrent RUNX1-RNA and RUNX1-DNA interactions, we annotated 24 185 high-confidence RUNX1-chromatin immunoprecipitation (ChIP) peaks to the same gene catalog and identified 13 275 corresponding gene loci (supplemental Figure 1D, right). The majority of these peaks were found at intronic, promoter, and intergenic regions (supplemental Figure 1F). Because most RUNX1-RIP and RUNX1-ChIP peaks were distributed at coding gene loci (Figure 1A-B), we focused our analyses on this gene group. By intersecting these genes with a list of 78 myeloid genes defined by their known roles in myeloid development, or as myeloid molecular markers (supplemental Table 1), we obtained 15 myeloid gene loci displaying both RUNX1-RIP and RUNX1-ChIP peaks (Figure 1C; supplemental Table 2). PU.1, a master regulator of myeloid development and a well-known transcriptional target of RUNX1,8 was among these genes. Intriguingly, we observed RNA peaks at the upstream region of PU.1 (Figure 1D). We further validated this observation by RUNX1 RIP-quantitative polymerase chain reaction (qPCR) (Figure 1E). Additional myeloid genes showing RUNX1-RIP peaks and RUNX1-ChIP peaks are presented in supplemental Figure 1G. The presence of previously uncharacterized RNAs, arising from the upstream region of the PU.1 locus, and able to interact with RUNX1, suggests their potential role in controlling PU.1 expression through RUNX1-mediated transcriptional regulation.

Screening of gene loci exhibiting concurrent RUNX1 RNA and DNA interactions in THP-1 cells. (A-B) Pie charts showing proportions of RUNX1 RIP-seq peaks and RUNX1 ChIP-seq peaks in coding and noncoding gene families. ChIP-seq data were from published source53 under the Gene Expression Omnibus (GEO) accession number GSE79899; snoRNA, small nucleolar RNAs; snRNA, small nuclear RNA; miRNA, microRNA; scaRNA, small cajal body-specific RNA. (C) Venn diagram intersecting RUNX1 RIP-seq and RUNX1 ChIP-seq gene lists and the myeloid gene list. (D) Gene track view of the PU.1 locus including the upstream region (highlighted in blue). Shown are RIP-seq tracks (Input, IgG, and RUNX1) and RUNX1 ChIP-seq tracks (GSM2108052). Data were integrated in the University of California, Santa Cruz (UCSC) genome browser. (E) RUNX1 RIP-qPCR confirmation. Left panel: location of 3 PCR amplicons (#1, #2, #3). Right panel: enrichment of RNAs captured by anti-RUNX1 antibody and IgG control at 3 amplicons relative to input. Error bars indicate SD (n = 3). **P < .01; ****P < .0001. See also supplemental Figure 1 and supplemental Table 3.
Screening of gene loci exhibiting concurrent RUNX1 RNA and DNA interactions in THP-1 cells. (A-B) Pie charts showing proportions of RUNX1 RIP-seq peaks and RUNX1 ChIP-seq peaks in coding and noncoding gene families. ChIP-seq data were from published source53 under the Gene Expression Omnibus (GEO) accession number GSE79899; snoRNA, small nucleolar RNAs; snRNA, small nuclear RNA; miRNA, microRNA; scaRNA, small cajal body-specific RNA. (C) Venn diagram intersecting RUNX1 RIP-seq and RUNX1 ChIP-seq gene lists and the myeloid gene list. (D) Gene track view of the PU.1 locus including the upstream region (highlighted in blue). Shown are RIP-seq tracks (Input, IgG, and RUNX1) and RUNX1 ChIP-seq tracks (GSM2108052). Data were integrated in the University of California, Santa Cruz (UCSC) genome browser. (E) RUNX1 RIP-qPCR confirmation. Left panel: location of 3 PCR amplicons (#1, #2, #3). Right panel: enrichment of RNAs captured by anti-RUNX1 antibody and IgG control at 3 amplicons relative to input. Error bars indicate SD (n = 3). **P < .01; ****P < .0001. See also supplemental Figure 1 and supplemental Table 3.
Characterization of the RUNX1-interacting lncRNA LOUP
To map the RUNX1-interacting transcript(s), we inspected the RNA expression and epigenetic landscape at the upstream region of the PU.1 locus (Figure 2A). Remarkably, the RNA-sequencing (RNA-seq) track view revealed 2 distinct RNA peaks. A narrow peak was observed at the URE, which corresponded to an area of open chromatin in myeloid cells as indicated by strong DNase I hypersensitivity signals (Figure 2A, DNase-seq [or DNase sequencing]). This element was also enriched with histone posttranslational modifications such as H3K27ac, H3K4me1, and H3K4me3 (Figure 2A, ChIP-seq [or ChIP sequencing]), which are typical features of active enhancers.31,32 A broad peak was proximal to the promoter region. Notably, these peaks were present in myeloid cell lines (THP-1 and HL-60) and primary monocytes, but not in the lymphoid cell line Jurkat, which does not express PU.1 messenger RNA (mRNA), indicating a cell-type–specific expression pattern. Reverse transcription polymerase chain reaction (RT-PCR) and Sanger sequencing analysis identified exon junctions connecting these 2 peaks in both human and murine cell lines (supplemental Figure 2A). Strand-specific RT-PCR analysis confirmed that the transcript is sense with respect to the PU.1 gene (Figure 2B). A strong Cap-analysis gene-expression sequencing (CAGE-seq) peak is present within the URE and in the sense genomic orientation (Figure 2A CAGE-seq), suggesting the presence of the 5′ end of a transcript. We further identified this 5′ end including a transcription start site (TSS) of the RNA within the homology region 1 (H1) of the URE18 (supplemental Figure 2B-C). Although a splicing event was detected within the second exon, intron retention was dominant as shown by the presence of a ∼2.3 kb major transcript and a ∼1.0 kb minor transcript (Figure 2C-D). The transcripts were detectable in the myeloid cell line U937, but not in the lymphoid cell line Jurkat, further indicating their cell-type specificity (Figure 2C). Notably, the RNA exhibited very low coding potential similar to that of other known lncRNAs (supplemental Figure 2E) as assessed by PhyloCSF software.33 Additionally, no known protein domains were found (data not shown) using PFAM software.34 Thus, we named the RNA transcript “long noncoding RNA originating from the URE of PU.1”, or LOUP. A LOUP homolog is also expressed and originated from the URE in murine cells (Ensembl ID #ENSMUST00000131400). Quantitative RT-PCR (qRT-PCR) analyses of subcellular fractionations revealed that it resides in both the cytoplasm and the nucleoplasm compartments, and was particularly enriched in the chromatin fraction (supplemental Figure 2F). The lncRNA is polyadenylated, being detected from oligo(dT)-primed complementary DNAs (cDNAs) (Figure 2B) and enriched in the polyA+ RNA fraction (Figure 2C-D; supplemental Figure 2G). LOUP is a low-abundant lncRNA; the spliced form is expressed as ∼40, 14, and 5 copies per cell in U937, HL-60, and NB4, respectively (Figure 2E). The lncRNA was barely detectable as its premature (nonspliced) form in total RNA as well as in the nuclear RNA fraction (supplemental Figure 2H-I). Altogether, these findings established LOUP as a polyadenylated lncRNA that emanates from the URE and extends toward the PrPr.
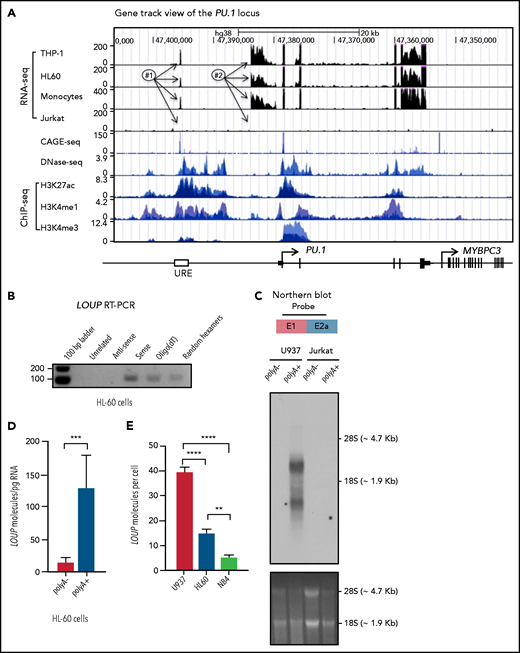
Characterization of long noncoding RNA LOUP. (A) Gene track view of the genomic region encompassing the PU.1 locus. RNA-seq tracks include THP-1, HL60, primary monocytes, and Jurkat. DNase-seq and ChIP-seq are overlay tracks of monocyte and myeloid cell lines. These data were processed from published data in GEO (see “Methods” for details). The CAGE-seq track was imported from the FANTOM5 project; #1, #2, and arrows point to locations of the RNA peaks. (B) RT-PCR analysis of LOUP’s transcript features. First-strand cDNAs were generated from HL-60 total RNA using a primer that does not anneal to the PU.1 locus (Unrelated), random hexamers, Oligo(dT), and strand-specific primers (antisense and sense). (C) Northern blot analysis of LOUP. polyA− and polyA+ RNA fractions were isolated from U937 and Jurkat cells. Top panel: schematic of the probe location spanning exon junction (E1 and E2a; see supplemental Figure 2D). Middle panel: northern blot detection of LOUP’s major and minor transcripts. Bottom panel: RNA gel demonstrating relative migration between 28S and 18S rRNAs stained with ethidium bromide. (D) qRT-PCR analysis of LOUP levels in polyA− and polyA+ RNA fractions isolated from HL-60 cells. Error bars indicate SD (n = 3). ***P < .001. (E) Calculation of LOUP transcript per cell by qRT-PCR. The LOUP RNA standard curve was generated by in vitro transcription. Error bars indicate SD (n = 3). **P < .01; ****P < .0001. See also supplemental Figure 2 and supplemental Table 3.
Characterization of long noncoding RNA LOUP. (A) Gene track view of the genomic region encompassing the PU.1 locus. RNA-seq tracks include THP-1, HL60, primary monocytes, and Jurkat. DNase-seq and ChIP-seq are overlay tracks of monocyte and myeloid cell lines. These data were processed from published data in GEO (see “Methods” for details). The CAGE-seq track was imported from the FANTOM5 project; #1, #2, and arrows point to locations of the RNA peaks. (B) RT-PCR analysis of LOUP’s transcript features. First-strand cDNAs were generated from HL-60 total RNA using a primer that does not anneal to the PU.1 locus (Unrelated), random hexamers, Oligo(dT), and strand-specific primers (antisense and sense). (C) Northern blot analysis of LOUP. polyA− and polyA+ RNA fractions were isolated from U937 and Jurkat cells. Top panel: schematic of the probe location spanning exon junction (E1 and E2a; see supplemental Figure 2D). Middle panel: northern blot detection of LOUP’s major and minor transcripts. Bottom panel: RNA gel demonstrating relative migration between 28S and 18S rRNAs stained with ethidium bromide. (D) qRT-PCR analysis of LOUP levels in polyA− and polyA+ RNA fractions isolated from HL-60 cells. Error bars indicate SD (n = 3). ***P < .001. (E) Calculation of LOUP transcript per cell by qRT-PCR. The LOUP RNA standard curve was generated by in vitro transcription. Error bars indicate SD (n = 3). **P < .01; ****P < .0001. See also supplemental Figure 2 and supplemental Table 3.
LOUP is a myeloid-specific lncRNA that is coexpressed with myeloid-lineage gene PU.1
We sought to explore LOUP expression in normal tissues and cell types. By examining the LOUP transcript profile in different human tissue types from the Illumina Body Map data set, we noticed that this lncRNA was barely detectable in most tissues but was elevated in leukocytes (Figure 3A). Remarkably, comparing with 2 of its closest neighbor genes, PU.1 and SLC39A13 (supplemental Figure 2D), the LOUP expression pattern was similar to that of PU.1 mRNA (Figure 3A-B) but not of SLC39A13 (supplemental Figure 3A). Additionally, LOUP transcript levels were not correlated with those of its interacting partner, RUNX1 (supplemental Figure 3B). To further delineate the relationship between LOUP and PU.1 transcript levels and lineage identity in individual blood cells, we used single-cell RNA (scRNA)-sequencing (scRNA-seq) analyses (supplemental Figure 3C). Notably, LOUP and PU.1 were both enriched in myeloid cells, comprising monocytes, macrophages, and granulocytes (supplemental Figure 3D-G). Expectedly, RUNX1 was broadly expressed in myeloid cells as well as lymphoid cells (T, B, and natural killer [NK]) (supplemental Figure 3H). By stratifying the mononuclear cell population into LOUP+/PU.1+ and LOUP -/PU.1− groups based on LOUP and PU.1 expression levels, we noted that LOUP−/PU.1− cells were associated with T, B, and NK cells. Remarkably, 99.3% of LOUP+/PU.1+ cells were linked to the myeloid identity (Figure 3C). Consistent with this observation, top biological processes associated with expression of LOUP and PU.1 were monocyte, macrophage, and granulocyte functions (supplemental Figure 3I; supplemental Table 4). We further examined expression patterns of LOUP and PU.1 during myeloid differentiation. qRT-PCR analyses of purified murine hematopoietic cell populations showed low Loup levels in long-term hematopoietic stem cells (LT-HSCs), short-term hematopoietic stem cells (ST-HSCs), common myeloid progenitors (CMPs), and megakaryocyte-erythroid progenitors (MEPs). Remarkably, Loup expression was elevated in myeloid progenitor cells (granulocyte-macrophage progenitors [GMPs]) and was highest in definitive myeloid cells (Figure 3D). A similar expression pattern was seen with PU.1 (Figure 3E). Taken together, our data indicated that LOUP and PU.1 transcript levels were associated with the myeloid identity.
![Expression profiles of LOUP and PU.1 in normal tissues and cell lineages. (A-B) Transcript profiles of LOUP and PU.1 in human tissues. Shown are transcript counts from the Illumina Body Map RNA-seq data set (ArrayExpress [AE]: E-MTAB-513). Error bars indicate SD (n = 2). (C) The proportion of cell lineages corresponding to transcript levels of LOUP and PU.1. scRNA-seq data of human mononuclear cells were retrieved from the 10x Genomic Project.54 Myeloid includes monocytes, macrophages, and granulocytes. Numbers on each bar indicate the percentage of each cell type within LOUP+/PU.1+ (left panel) and LOUP−/PU.1− groups (right panel), respectively. (D-E) qRT-PCR analysis of Loup RNA and Pu.1 mRNA levels in murine hematopoietic stem, progenitor, and mature (myeloid) cell populations. Data are shown relative to LT-HSCs. Error bars indicate SD (n = 2). See also supplemental Figure 3 and supplemental Table 3. B, B lymphocyte; CMP, common myeloid progenitor; DC, dendritic cell; Ery, erythrocyte; GMP, granulocyte-macrophage progenitor, myeloid cells (Mac1+Gr1+); LMPP, lymphoid-primed multipotent progenitor; LT-HSC, long-term hematopoietic stem cell; Meg, megakaryocyte; MEP, megakaryocyte-erythroid progenitor; NK, natural killer cell; Plas, plasma cell; ST-HSC, short-term hematopoietic stem cell; TCD4+, T helper cell; TCD8+, cytotoxic T cell; Treg, regulatory T cell.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/138/15/10.1182_blood.2020007920/6/m_bloodbld2020007920f3.png?Expires=1765904598&Signature=jdOvENv~QIo1A9WUXvucCp5XxsLxZawriyBqbThyCGeovvT~eVsl2Yl-7RW8OjNDa6a1YbyBA5rxDy57pGy3Le0ogd9qdmyjDfyFyg5FwC7NABwZ3yxZgUX2F1ZPzjf2gfiJdLbqH9Nk3T8mLkCh8EWIamZIYAyh61ObDj8L0id5KCcv142Uu3jD8RKCc1U3uX5RSalfYJspl1L7egie-G1XyR61HH2ZBaViE1G5SRIQt5bc2Ia2AqD1ueTjKx7j0lAbCiLMe0V7guAx7AY2Zg-mHw3vVfCL5d2hpaEWQlswsE7TyNd~UvHy-p4FiWk4j-jjnd4~XhcQbTCBguvgkw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Expression profiles of LOUP and PU.1 in normal tissues and cell lineages. (A-B) Transcript profiles of LOUP and PU.1 in human tissues. Shown are transcript counts from the Illumina Body Map RNA-seq data set (ArrayExpress [AE]: E-MTAB-513). Error bars indicate SD (n = 2). (C) The proportion of cell lineages corresponding to transcript levels of LOUP and PU.1. scRNA-seq data of human mononuclear cells were retrieved from the 10x Genomic Project.54 Myeloid includes monocytes, macrophages, and granulocytes. Numbers on each bar indicate the percentage of each cell type within LOUP+/PU.1+ (left panel) and LOUP−/PU.1− groups (right panel), respectively. (D-E) qRT-PCR analysis of Loup RNA and Pu.1 mRNA levels in murine hematopoietic stem, progenitor, and mature (myeloid) cell populations. Data are shown relative to LT-HSCs. Error bars indicate SD (n = 2). See also supplemental Figure 3 and supplemental Table 3. B, B lymphocyte; CMP, common myeloid progenitor; DC, dendritic cell; Ery, erythrocyte; GMP, granulocyte-macrophage progenitor, myeloid cells (Mac1+Gr1+); LMPP, lymphoid-primed multipotent progenitor; LT-HSC, long-term hematopoietic stem cell; Meg, megakaryocyte; MEP, megakaryocyte-erythroid progenitor; NK, natural killer cell; Plas, plasma cell; ST-HSC, short-term hematopoietic stem cell; TCD4+, T helper cell; TCD8+, cytotoxic T cell; Treg, regulatory T cell.
Expression profiles of LOUP and PU.1 in normal tissues and cell lineages. (A-B) Transcript profiles of LOUP and PU.1 in human tissues. Shown are transcript counts from the Illumina Body Map RNA-seq data set (ArrayExpress [AE]: E-MTAB-513). Error bars indicate SD (n = 2). (C) The proportion of cell lineages corresponding to transcript levels of LOUP and PU.1. scRNA-seq data of human mononuclear cells were retrieved from the 10x Genomic Project.54 Myeloid includes monocytes, macrophages, and granulocytes. Numbers on each bar indicate the percentage of each cell type within LOUP+/PU.1+ (left panel) and LOUP−/PU.1− groups (right panel), respectively. (D-E) qRT-PCR analysis of Loup RNA and Pu.1 mRNA levels in murine hematopoietic stem, progenitor, and mature (myeloid) cell populations. Data are shown relative to LT-HSCs. Error bars indicate SD (n = 2). See also supplemental Figure 3 and supplemental Table 3. B, B lymphocyte; CMP, common myeloid progenitor; DC, dendritic cell; Ery, erythrocyte; GMP, granulocyte-macrophage progenitor, myeloid cells (Mac1+Gr1+); LMPP, lymphoid-primed multipotent progenitor; LT-HSC, long-term hematopoietic stem cell; Meg, megakaryocyte; MEP, megakaryocyte-erythroid progenitor; NK, natural killer cell; Plas, plasma cell; ST-HSC, short-term hematopoietic stem cell; TCD4+, T helper cell; TCD8+, cytotoxic T cell; Treg, regulatory T cell.
LOUP induces PU.1 expression, promotes myeloid differentiation, and inhibits cell growth
To test our hypothesis that LOUP induces PU.1 expression, we first investigated the impact of loss-of-function of LOUP on PU.1 expression. Depletion of LOUP in macrophage cell line U937, which expresses a high level of LOUP (Figure 2E), via clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein 9 (Cas9) technology, resulted in a significant reduction in PU.1 mRNA levels (Figure 4A; supplemental Figure 4A-E). In line with this finding, knockdown of LOUP by short hairpin RNAs (shRNAs) in CD34+ hematopoietic stem/progenitor cells (HSPCs) cultured with myeloid differentiation-promoting cytokines, including interleukin 3 (IL-3), granulocyte-macrophage colony-stimulating factor (GM-CSF), and granulocyte colony-stimulating factor (G-CSF), resulted in PU.1 reduction (Figure 4B). In gain-of-function experiments, in trans-overexpression of LOUP resulted in significant induction of PU.1 (supplemental Figure 4F-G). Remarkably, in cis locus-specific induction of endogenous LOUP via the CRISPR/dCas9-VP64 activation system yielded a comparable increase in PU.1 expression (Figure 4C; supplemental Figure 4A). LOUP overexpression was associated with a decrease in total cell number (Figure 4D), whereas LOUP depletion was associated with increased cell proliferation (supplemental Figure 4H), suggesting that LOUP inhibits cell growth. Consistent with the important role of PU.1 in myeloid differentiation,6,35,36LOUP depletion by either shRNAs or CRISPR/Cas9 technology was associated with a reduction in expression of the myeloid marker CD11b (Figure 4E; supplemental Figure 4I), whereas LOUP overexpression resulted in an increase in CD11b levels (supplemental Figure 4J). Additionally, LOUP knockdown in CD34+ HSPCs resulted in cells with less-differentiated morphology and a reduction in the expression level of the macrophage marker CD14 upon myeloid induction (Figure 4F; supplemental Figure 4K). Together, these results demonstrate that LOUP promotes myeloid differentiation and inhibits cell growth.
![The effect of LOUP on PU.1 expression, myeloid differentiation, and cell growth. (A-C) qRT-PCR expression analysis for LOUP (left panels) and PU.1 (right panels). (A) Nontargeting (N) and LOUP-targeting (L) U937 CRISPR/Cas9 cell clones. Data are shown relative to N1 control. (B) Cord blood CD34+ HSPCs stably transfected with LOUP-targeting (shLOUP) or nontargeting (shControl) shRNAs by lentiviral transduction and grown in liquid culture with myeloid differentiation-promoting cytokines for 12 days. (C) K562 dCas9-VP64-stable cells infected with LOUP-targeting (#A1 and #A2) or nontargeting (Control) sgRNAs. (D) Trypan blue exclusion and manual cell counts for kinetics of cell growth (sgControl vs sgLOUP [#A1 and #A2]). (E) Representative flow cytometry results of CD11b myeloid marker expression. Cord blood CD34+ HSPCs stably transfected with either shLOUP shRNA or shControl shRNA by lentiviral transduction and grown in liquid culture with myeloid differentiation-promoting cytokines for 12 days. (F) Camco Stain Pak staining of shControl and shLOUP samples as described in panel E. Representative images were acquired with a Nikon Eclipse microscope (original magnification, ×60) and the SPOT Insight2 camera. Error bars indicate SD (n = 3). *P < .05; **P < .01; ***P < .001; ****P < .0001. See also supplemental Figure 4 and supplemental Table 3. n.s., not significant.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/138/15/10.1182_blood.2020007920/6/m_bloodbld2020007920f4.png?Expires=1765904598&Signature=G6V~4BIEPqyLtUZ2~2OV1ECdPR5BEHglCRyGM85VJ~5QwYdQss2QOdwb-c-ReRQFqSDhTu49Mm4neWgTnGeCgPQIzGuydlsHyy6KwBqLvHoUQslLdbgkBwNH2rsyek5FHRmTz4PCDjx4EhabK-cTsM6NHuZUrW9NDuFF4TgjH2bfqAxmQr9TdOiMvyAPhFPFtjUm7X2tBmBW6KNIfnTUmfBFEaEPtImEMOZgFQUaHf3XpFAI3iuLI79zpozjj3tYJbd1zQPeKDaHwzC8llcXiu91XTJoEzAlHUcp~5IkiQhJlVkH-xKoHN8Z6cMkQg24ZmJSWSPGLlKjpiDapWM5qQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
The effect of LOUP on PU.1 expression, myeloid differentiation, and cell growth. (A-C) qRT-PCR expression analysis for LOUP (left panels) and PU.1 (right panels). (A) Nontargeting (N) and LOUP-targeting (L) U937 CRISPR/Cas9 cell clones. Data are shown relative to N1 control. (B) Cord blood CD34+ HSPCs stably transfected with LOUP-targeting (shLOUP) or nontargeting (shControl) shRNAs by lentiviral transduction and grown in liquid culture with myeloid differentiation-promoting cytokines for 12 days. (C) K562 dCas9-VP64-stable cells infected with LOUP-targeting (#A1 and #A2) or nontargeting (Control) sgRNAs. (D) Trypan blue exclusion and manual cell counts for kinetics of cell growth (sgControl vs sgLOUP [#A1 and #A2]). (E) Representative flow cytometry results of CD11b myeloid marker expression. Cord blood CD34+ HSPCs stably transfected with either shLOUP shRNA or shControl shRNA by lentiviral transduction and grown in liquid culture with myeloid differentiation-promoting cytokines for 12 days. (F) Camco Stain Pak staining of shControl and shLOUP samples as described in panel E. Representative images were acquired with a Nikon Eclipse microscope (original magnification, ×60) and the SPOT Insight2 camera. Error bars indicate SD (n = 3). *P < .05; **P < .01; ***P < .001; ****P < .0001. See also supplemental Figure 4 and supplemental Table 3. n.s., not significant.
The effect of LOUP on PU.1 expression, myeloid differentiation, and cell growth. (A-C) qRT-PCR expression analysis for LOUP (left panels) and PU.1 (right panels). (A) Nontargeting (N) and LOUP-targeting (L) U937 CRISPR/Cas9 cell clones. Data are shown relative to N1 control. (B) Cord blood CD34+ HSPCs stably transfected with LOUP-targeting (shLOUP) or nontargeting (shControl) shRNAs by lentiviral transduction and grown in liquid culture with myeloid differentiation-promoting cytokines for 12 days. (C) K562 dCas9-VP64-stable cells infected with LOUP-targeting (#A1 and #A2) or nontargeting (Control) sgRNAs. (D) Trypan blue exclusion and manual cell counts for kinetics of cell growth (sgControl vs sgLOUP [#A1 and #A2]). (E) Representative flow cytometry results of CD11b myeloid marker expression. Cord blood CD34+ HSPCs stably transfected with either shLOUP shRNA or shControl shRNA by lentiviral transduction and grown in liquid culture with myeloid differentiation-promoting cytokines for 12 days. (F) Camco Stain Pak staining of shControl and shLOUP samples as described in panel E. Representative images were acquired with a Nikon Eclipse microscope (original magnification, ×60) and the SPOT Insight2 camera. Error bars indicate SD (n = 3). *P < .05; **P < .01; ***P < .001; ****P < .0001. See also supplemental Figure 4 and supplemental Table 3. n.s., not significant.
LOUP induces enhancer-promoter communication by interacting with chromatin at the PU.1 locus
We have previously reported that the formation of a chromatin loop mediated by URE-PrPr interaction is crucial for PU.1 induction.18,19 Because LOUP arises from the URE and extends toward the PrPr, we reasoned that LOUP drives long-range transcription of PU.1 by promoting URE-PrPr interaction. To elucidate this, we quantified interaction strengths of the URE with the PrPr and the surrounding area by chromosome conformation capture and TaqMan qPCR (3C-qPCR) (Figure 5A). Consistent with previous reports,18,19 we detected strong interaction between the URE and the PrPr, but not between the URE and other genomic regions, including the upstream PU.1 promoter, intergenic sequences, and the MYBPC3 gene body downstream of the PU.1 locus. Interestingly, LOUP depletion caused a significant reduction in URE-PrPr communication (Figure 5B). To provide evidence supporting our prediction that LOUP recruits the URE to the PrPr by physically interacting with the 2 elements, we used the chromatin isolation by RNA purification (ChIRP) assay.37 Biotinylated LOUP-tiling oligos were able to capture endogenous LOUP RNA in U937 cells (Figure 5C). Enrichment of the URE and the PrPr cocaptured with LOUP RNA was observed in ChIRP-ed samples with LOUP-tiling probes but not LacZ-tiling controls, suggesting that LOUP occupies both the URE and the PrPr (Figure 5D). Taken together, our data indicate that by interacting and bringing to close proximity 2 regulatory elements, the URE and the PrPr, LOUP promotes the formation of a functional chromatin loop within the PU.1 locus that is critical in inducing PU.1 expression.
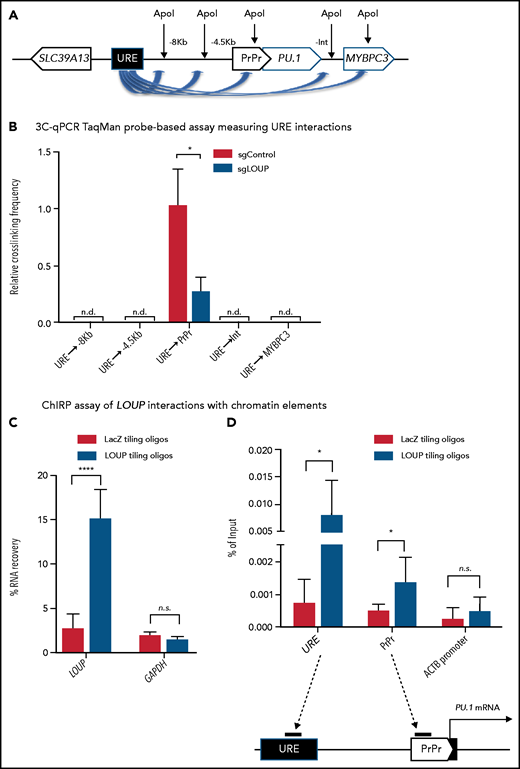
3C and ChIRP assays measuring the effect of LOUP on chromatin looping. (A) Schematic diagram illustrating potential 3C interactions between the URE and genomic viewpoints surrounding the PU.1 locus. Included are restriction recognition sites of ApoI used in the assay. -8 kb and -4 kb are distances from the PrPr in kilobases. (B) 3C-qPCR TaqMan probe-based assay comparing cross-linking frequencies at chromatin viewpoints. The U937 cell clone L2a, carrying a LOUP-homozygous indel that does not alter the recognition pattern of ApoI (supplemental Figure 4D), was used to compare with nontargeting control (sgControl, N1). (C) qRT-PCR assay evaluating levels of LOUP RNA and control GAPDH captured by biotinylated LOUP-tiling and LacZ-tiling probes using ChIRP. (D) ChIRP assay assessing LOUP occupancies at the URE, the PrPr, and ACTB promoter. LOUP-tiling oligos were used to capture endogenous LOUP in U937 cells. LacZ-tiling oligos were used as negative control. Error bars indicate SD (n = 3). *P < .05; ****P < .0001. Int, intergenic; n.d., not detectable.
3C and ChIRP assays measuring the effect of LOUP on chromatin looping. (A) Schematic diagram illustrating potential 3C interactions between the URE and genomic viewpoints surrounding the PU.1 locus. Included are restriction recognition sites of ApoI used in the assay. -8 kb and -4 kb are distances from the PrPr in kilobases. (B) 3C-qPCR TaqMan probe-based assay comparing cross-linking frequencies at chromatin viewpoints. The U937 cell clone L2a, carrying a LOUP-homozygous indel that does not alter the recognition pattern of ApoI (supplemental Figure 4D), was used to compare with nontargeting control (sgControl, N1). (C) qRT-PCR assay evaluating levels of LOUP RNA and control GAPDH captured by biotinylated LOUP-tiling and LacZ-tiling probes using ChIRP. (D) ChIRP assay assessing LOUP occupancies at the URE, the PrPr, and ACTB promoter. LOUP-tiling oligos were used to capture endogenous LOUP in U937 cells. LacZ-tiling oligos were used as negative control. Error bars indicate SD (n = 3). *P < .05; ****P < .0001. Int, intergenic; n.d., not detectable.
LOUP binds the Runt domain of RUNX1 and coordinates recruitment of RUNX1 to the enhancer and the promoter
Because LOUP interacts with RUNX1 (Figure 1), we asked whether LOUP mediates the URE-PrPr interaction by cooperating with RUNX1. In line with the previous finding in murine cells,15 we observed RUNX1 occupancy at the URE in primary CD34+ cells isolated from healthy donors and patients with AML. Importantly, we noticed a peak at the PrPr, indicating that RUNX1 also occupies the PrPr (Figure 6A). We further inspected the genomic region surrounding the PrPr and found a RUNX-DNA–binding consensus motif at -220 bp relative to the PU.1 mRNA transcription start site. Biotinylated DNA pulldown (DNAP) assays demonstrated that probes, containing the RUNX consensus motifs embedded in the URE and the PrPr, efficiently captured endogenous RUNX1 from U937 nuclear extracts (Figure 6B; supplemental Figure 5A), suggesting that RUNX1 binds its DNA consensus motif at both the URE and the PrPr. Furthermore, our ChIP-qPCR data revealed that RUNX1 occupancy at both the URE and the PrPr was reduced upon LOUP depletion (Figure 6C) but increased following LOUP induction (supplemental Figure 5B), indicating that LOUP promotes placement of RUNX1 dimers at the URE and the PrPr. Besides, LOUP depletion was associated with reduced C/EBPα occupancy (supplemental Figure 5C), but had no effect on PU.1 occupancy in the URE (supplemental Figure 5D), suggesting the involvement of LOUP in C/EBPα recruitment.
![LOUP cooperates with RUNX1 to facilitate URE-PrPr interaction. (A) Gene track view of the ∼26-kb region encompassing the URE and the PrPr. Top panel: RUNX1 ChIP-seq tracks derived from CD34+ cells from healthy donors (GSM1097884), and a patient with FLT3-ITD AML (GSM1581788). Bottom panel: a schematic showing the corresponding genomic locations of LOUP and the 5′ region of PU.1. (B) DNA pulldown assay showing binding of RUNX1 to the RUNX1-binding motifs at the URE and the PrPr. Proteins captured by biotinylated DNA oligos (wild-type oligo containing RUNX1-binding motif [wt]; oligo with mutated RUNX1-binding motif [mt]) in U937 nuclear lysate were detected by immunoblot. (C) ChIP-qPCR analysis of RUNX1 occupancy at the URE and the PrPr. LOUP-depleted U937 (sgLOUP, L2a) and control (sgControl, N1) clones were used. PCR amplicons include the URE (contains known RUNX1-binding motif at the URE), PrPr (contains putative RUNX1-binding motif in the PrPr), and GENE DESERT (a genome region that is devoid of protein-coding genes). Error bars indicate SD (n = 3). **P < .01; ****P < .0001. (D) RNA pulldown analysis of the RUNX1-LOUP interaction. Top panel: schematic diagram of LOUP showing the relative position of the repetitive region (RR). Arrows underneath the diagram illustrate direction and relative lengths of in vitro–transcribed and biotin-labeled LOUP fragments (AS, full-length antisense control; Bead, no RNA control; EGFP, EGFP mRNA control; RR; S, full-length sense). Bottom panel: LOUP fragments were incubated with U937 nuclear lysate. Retrieved proteins were identified by immunoblot. (E) Schematic diagram of the repetitive region (RR) showing predicted binding regions R1 and R2. (F-G) RNAP-binding analysis of R1 and R2 with recombinant full-length and Runt domain of RUNX1. In vitro-transcribed and biotin-labeled RNAs include R1-AS (R1 antisense control); R1-S (R1 sense); and R2-S (R2 sense). The vertical line demarcates where an unrelated lane was removed from the figure. See also supplemental Figure 5.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/138/15/10.1182_blood.2020007920/6/m_bloodbld2020007920f6.png?Expires=1765904598&Signature=sAaexxd9uqzTrkffHaV5AIP97knz~PZqVl97vMbKFrc~OuGfuMLUocTaXyX7ERSYoWA61vuTumR4nJmxElL1OmtfPzM3T1UYNYJYSUSGnrAULtlAJ6ATEo-mkV42yCcHRZ2pyPQw~oUCl2OmbpA-NxlONu5NCaOk8fbNrO7uVciNZhrHYIa46MXe~5FnBiQijOjpKLWPWEgxggBFY6l9XjibAf9f3opGRpop2OA4Iq8-u8WQxgmnDesG81sK6niyex2UuNJZbMDkeGdXwEzF~27KZMS~n0dphW1SiBVa~8fjWRL9Y3daAAjFRLVdDaeX6PPHIANIPz8mCCPW1STfWw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
LOUP cooperates with RUNX1 to facilitate URE-PrPr interaction. (A) Gene track view of the ∼26-kb region encompassing the URE and the PrPr. Top panel: RUNX1 ChIP-seq tracks derived from CD34+ cells from healthy donors (GSM1097884), and a patient with FLT3-ITD AML (GSM1581788). Bottom panel: a schematic showing the corresponding genomic locations of LOUP and the 5′ region of PU.1. (B) DNA pulldown assay showing binding of RUNX1 to the RUNX1-binding motifs at the URE and the PrPr. Proteins captured by biotinylated DNA oligos (wild-type oligo containing RUNX1-binding motif [wt]; oligo with mutated RUNX1-binding motif [mt]) in U937 nuclear lysate were detected by immunoblot. (C) ChIP-qPCR analysis of RUNX1 occupancy at the URE and the PrPr. LOUP-depleted U937 (sgLOUP, L2a) and control (sgControl, N1) clones were used. PCR amplicons include the URE (contains known RUNX1-binding motif at the URE), PrPr (contains putative RUNX1-binding motif in the PrPr), and GENE DESERT (a genome region that is devoid of protein-coding genes). Error bars indicate SD (n = 3). **P < .01; ****P < .0001. (D) RNA pulldown analysis of the RUNX1-LOUP interaction. Top panel: schematic diagram of LOUP showing the relative position of the repetitive region (RR). Arrows underneath the diagram illustrate direction and relative lengths of in vitro–transcribed and biotin-labeled LOUP fragments (AS, full-length antisense control; Bead, no RNA control; EGFP, EGFP mRNA control; RR; S, full-length sense). Bottom panel: LOUP fragments were incubated with U937 nuclear lysate. Retrieved proteins were identified by immunoblot. (E) Schematic diagram of the repetitive region (RR) showing predicted binding regions R1 and R2. (F-G) RNAP-binding analysis of R1 and R2 with recombinant full-length and Runt domain of RUNX1. In vitro-transcribed and biotin-labeled RNAs include R1-AS (R1 antisense control); R1-S (R1 sense); and R2-S (R2 sense). The vertical line demarcates where an unrelated lane was removed from the figure. See also supplemental Figure 5.
LOUP cooperates with RUNX1 to facilitate URE-PrPr interaction. (A) Gene track view of the ∼26-kb region encompassing the URE and the PrPr. Top panel: RUNX1 ChIP-seq tracks derived from CD34+ cells from healthy donors (GSM1097884), and a patient with FLT3-ITD AML (GSM1581788). Bottom panel: a schematic showing the corresponding genomic locations of LOUP and the 5′ region of PU.1. (B) DNA pulldown assay showing binding of RUNX1 to the RUNX1-binding motifs at the URE and the PrPr. Proteins captured by biotinylated DNA oligos (wild-type oligo containing RUNX1-binding motif [wt]; oligo with mutated RUNX1-binding motif [mt]) in U937 nuclear lysate were detected by immunoblot. (C) ChIP-qPCR analysis of RUNX1 occupancy at the URE and the PrPr. LOUP-depleted U937 (sgLOUP, L2a) and control (sgControl, N1) clones were used. PCR amplicons include the URE (contains known RUNX1-binding motif at the URE), PrPr (contains putative RUNX1-binding motif in the PrPr), and GENE DESERT (a genome region that is devoid of protein-coding genes). Error bars indicate SD (n = 3). **P < .01; ****P < .0001. (D) RNA pulldown analysis of the RUNX1-LOUP interaction. Top panel: schematic diagram of LOUP showing the relative position of the repetitive region (RR). Arrows underneath the diagram illustrate direction and relative lengths of in vitro–transcribed and biotin-labeled LOUP fragments (AS, full-length antisense control; Bead, no RNA control; EGFP, EGFP mRNA control; RR; S, full-length sense). Bottom panel: LOUP fragments were incubated with U937 nuclear lysate. Retrieved proteins were identified by immunoblot. (E) Schematic diagram of the repetitive region (RR) showing predicted binding regions R1 and R2. (F-G) RNAP-binding analysis of R1 and R2 with recombinant full-length and Runt domain of RUNX1. In vitro-transcribed and biotin-labeled RNAs include R1-AS (R1 antisense control); R1-S (R1 sense); and R2-S (R2 sense). The vertical line demarcates where an unrelated lane was removed from the figure. See also supplemental Figure 5.
By aligning the LOUP sequence with itself using the Basic Local Alignment Search Tool (BLAST), we unexpectedly uncovered a highly repetitive region (RR) of 670 bp near the 3′ end of LOUP (supplemental Figure 5E). Interestingly, by performing RNA pulldown (RNAP) assays, we noted that biotinylated LOUP RR was able to capture endogenous RUNX1 proteins in U937 nuclear extracts at a level that was comparable to biotinylated full-length LOUP, indicating that the RR contains the RUNX1-binding region (Figure 6D; supplemental Figure 5F). To further locate the binding region, we first computed the potential interaction strength of putative elements within the RR to RUNX1 protein by using the catRAPID algorithm.38 By doing so, we identified 2 ∼100-bp candidate regions, termed region 1 (R1) and region 2 (R2), with high interaction scores (Figure 6E; supplemental Figure 5G). We also identified a repertoire of RIP peaks containing sequences similar to that of R1 or R2, indicating that these regions might represent general RUNX1-binding motifs (supplemental Tables 5 and 6). RNAP analysis confirmed that R1 and R2 bind recombinant RUNX1 (Figure 6F; supplemental Figure 5H). Additionally, the recombinant Runt domain of RUNX1 was able to bind R1 and R2 (Figure 6G; supplemental Figure 5I), suggesting that the Runt domain is responsible for LOUP binding. These data together suggest that LOUP binds RUNX1 and coordinates deposition of RUNX1 dimers to the URE and the PrPr.
RUNX1-ETO downregulates LOUP in t(8;21) AML by inhibiting histone H3 acetylation and reducing chromatin accessibility at the URE
We further examined how the oncogenic fusion protein RUNX1-ETO, derived from t(8;21) chromosomal translocation, affects the regulatory function of LOUP. By examining LOUP transcript profiles in an AML RNA-seq data set downloaded from The Cancer Genome Atlas (TCGA), we noticed that LOUP RNA levels were significantly lower in t(8;21) patients with AML as compared with AML patients with normal karyotype (supplemental Figure 6A left panel). Consistent with our data demonstrating that PU.1 is a downstream target of LOUP, PU.1 levels were also lower in t(8;21) patients with AML (supplemental Figure 6A right panel). These findings were further confirmed by qRT-PCR using patient samples (Figure 7A). Additionally, we found a robust correlation between LOUP and PU.1 RNA levels (supplemental Figure 6B). Thus, we reasoned that LOUP may act as an inhibitory target of RUNX1-ETO in t(8;21) AML. Indeed, RUNX1-ETO overexpression repressed LOUP and PU.1 expression (Figure 7B; supplemental Figure 6C-D). Importantly, enforced LOUP expression rescues RUNX1-ETO’s effect on PU.1 inhibition (Figure 7B), indicating LOUP-dependent induction of PU.1. Conversely, depletion of RUNX1-ETO in t(8;21) AML cell Kasumi-1 by either small interfering RNAs (siRNAs) or shRNAs resulted in a robust increase in LOUP transcript levels, accompanied by a significant induction in PU.1 mRNA (supplemental Figure 6E-F). RUNX1-ETO is capable of recruiting the nuclear receptor corepressor histone deacetylase complex and associates with histone deacetylase activity.39-41 To examine whether RUNX1-ETO inhibits LOUP transcription by affecting local histone acetylation, we analyzed histone acetylation and chromatin accessibility at the URE, where LOUP transcription is initiated, upon depletion of RUNX1-ETO.42 As expected, knockdown of RUNX1-ETO reduces RUNX1-ETO occupancy at the URE (Figure 7C, top panel). Interestingly, depletion of RUNX1-ETO resulted in a robust induction of the H3K9Ac histone acetylation mark (Figure 7C, middle panel; supplemental Figure 6G), and DNase I accessibility at the URE (Figure 7C, bottom panel). We further confirmed that RUNX1-ETO binds the RUNX1 sites in the URE (Figure 7D; supplemental Figure 6H). Taken together, our data indicate that RUNX1-ETO exerts its opposing regulatory effect on PU.1 expression, at least in part, by inhibiting LOUP transcription through reducing URE accessibility.
![Effects of RUNX1-ETO on regulatory function of LOUP. (A) qRT-PCR analysis of samples from patients with AML. Normal karyotype (NK), n = 14; t(8;21) karyotype, n = 7. Mann-Whitney U test: **P < .01; *P < .05. (B) qRT-PCR expression analysis of LOUP RNA (left panel) and PU.1 mRNA (right panel) in U937 cells with inducible expression of RUNX1-ETO (+/−, with/without induction). Cells were transfected with empty vector (EV) and LOUP cDNA. Error bars indicate SD (n = 3). *P < .05; **P < .01. (C) Gene track view at the LOUP locus including the URE where LOUP transcription initiation is located. From top to bottom are RUNX1-ETO ChIP-seq tracks, H3K9Ac ChIP-seq tracks, and DNase-seq tracks of Kasumi-1 cells upon depletion of RUNX1-ETO. Cells were transfected with either nontargeting siRNA (siControl) or RUNX1-ETO–targerting siRNA. Data were processed from a published data set (GEO GSE29222) and integrated into the UCSC genome browser. (D) DNA pulldown assay showing binding of RUNX1-ETO to the RUNX1-binding motifs at the URE. Proteins captured by biotinylated DNA oligos (wild-type oligo containing RUNX1-binding motif [wt]; oligo with mutated RUNX1-binding motif [mt]) in Kasumi-1 nuclear lysate were detected by immunoblot (cytosol fraction [cyt]; nuclear fraction [nuc]). (E) A model of how LOUP coordinates with RUNX1 to modulate chromatin looping resulting in PU.1 induction, myeloid differentiation, and cell growth, and how RUNX1-ETO interferes with LOUP-mediated molecular functions.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/138/15/10.1182_blood.2020007920/6/m_bloodbld2020007920f7.png?Expires=1765904598&Signature=WqRlZ9iC-wkOCiE1XRbSvOOIoCPtfUVDfuTGfVhpwNsgKJR88r1OGgrvWSqLvISTF5KVT5M9R83NOyEk5Hyyso6~wz2up3Htir1ryFAVMsFWaMKBYebhrCVKLoYWKaAXDbF1Uh~9l5iabDwFhUpRxLabHoCzPk9nK5ApIMNzoOPbnSkMqx41Ww6Ltud7xSwKt544ClSRsPGzztD2BVdA14ikSUQz46HXp9Vs1spyBJRIM0DXIivgvHyRftkArn3WI~~ItRhjWISQTFnBN5m7utsRcfE9UtLTGnGRFb3Q3jCcvocdcfXGFXwIGCal92fOT1jK02PWFDtCMCrseE46-Q__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Effects of RUNX1-ETO on regulatory function of LOUP. (A) qRT-PCR analysis of samples from patients with AML. Normal karyotype (NK), n = 14; t(8;21) karyotype, n = 7. Mann-Whitney U test: **P < .01; *P < .05. (B) qRT-PCR expression analysis of LOUP RNA (left panel) and PU.1 mRNA (right panel) in U937 cells with inducible expression of RUNX1-ETO (+/−, with/without induction). Cells were transfected with empty vector (EV) and LOUP cDNA. Error bars indicate SD (n = 3). *P < .05; **P < .01. (C) Gene track view at the LOUP locus including the URE where LOUP transcription initiation is located. From top to bottom are RUNX1-ETO ChIP-seq tracks, H3K9Ac ChIP-seq tracks, and DNase-seq tracks of Kasumi-1 cells upon depletion of RUNX1-ETO. Cells were transfected with either nontargeting siRNA (siControl) or RUNX1-ETO–targerting siRNA. Data were processed from a published data set (GEO GSE29222) and integrated into the UCSC genome browser. (D) DNA pulldown assay showing binding of RUNX1-ETO to the RUNX1-binding motifs at the URE. Proteins captured by biotinylated DNA oligos (wild-type oligo containing RUNX1-binding motif [wt]; oligo with mutated RUNX1-binding motif [mt]) in Kasumi-1 nuclear lysate were detected by immunoblot (cytosol fraction [cyt]; nuclear fraction [nuc]). (E) A model of how LOUP coordinates with RUNX1 to modulate chromatin looping resulting in PU.1 induction, myeloid differentiation, and cell growth, and how RUNX1-ETO interferes with LOUP-mediated molecular functions.
Effects of RUNX1-ETO on regulatory function of LOUP. (A) qRT-PCR analysis of samples from patients with AML. Normal karyotype (NK), n = 14; t(8;21) karyotype, n = 7. Mann-Whitney U test: **P < .01; *P < .05. (B) qRT-PCR expression analysis of LOUP RNA (left panel) and PU.1 mRNA (right panel) in U937 cells with inducible expression of RUNX1-ETO (+/−, with/without induction). Cells were transfected with empty vector (EV) and LOUP cDNA. Error bars indicate SD (n = 3). *P < .05; **P < .01. (C) Gene track view at the LOUP locus including the URE where LOUP transcription initiation is located. From top to bottom are RUNX1-ETO ChIP-seq tracks, H3K9Ac ChIP-seq tracks, and DNase-seq tracks of Kasumi-1 cells upon depletion of RUNX1-ETO. Cells were transfected with either nontargeting siRNA (siControl) or RUNX1-ETO–targerting siRNA. Data were processed from a published data set (GEO GSE29222) and integrated into the UCSC genome browser. (D) DNA pulldown assay showing binding of RUNX1-ETO to the RUNX1-binding motifs at the URE. Proteins captured by biotinylated DNA oligos (wild-type oligo containing RUNX1-binding motif [wt]; oligo with mutated RUNX1-binding motif [mt]) in Kasumi-1 nuclear lysate were detected by immunoblot (cytosol fraction [cyt]; nuclear fraction [nuc]). (E) A model of how LOUP coordinates with RUNX1 to modulate chromatin looping resulting in PU.1 induction, myeloid differentiation, and cell growth, and how RUNX1-ETO interferes with LOUP-mediated molecular functions.
In summary, we established LOUP as a myeloid-specific lncRNA that promotes myeloid differentiation and inhibits cell growth via cooperating with RUNX1 to induce PU.1 expression, and also established that RUNX1-ETO disrupts the action of LOUP in t(8;21) AML. Thus, lncRNA LOUP acts as a regulatory hub delivering opposing effects from a broadly expressed transcription factor and its oncogenic derivative on long-range transcription of an important lineage gene (Figure 7E).
Discussion
In this study, we discovered that RUNX1, which is expressed and exerts its regulatory roles in diverse cell types,43,44 cooperates with myeloid-specific lncRNA LOUP to induce long-range transcription of PU.1, and that RUNX1-ETO impairs LOUP-mediated PU.1 induction by inhibiting LOUP expression in t(8;21) AML. Our study reported several important mechanistic findings. We reveal LOUP as a cellular RNA-interacting partner of RUNX1. We also demonstrate that LOUP recruits RUNX1 to respective RUNX1-binding motifs at both the URE and the PrPr, thereby promoting formation of the URE-PrPr chromatin loop at the PU.1 locus. Additionally, we identify a repetitive region serving as the RUNX1-binding platform for LOUP. Furthermore, we show that LOUP is an inhibitory target of RUNX1-ETO in t(8;21) AML. These findings provide important insight into how long-range transcription is induced in a gene-specific manner by ubiquitous transcription factors and how their chimeric derivatives disrupt normal gene induction in leukemia.
Our findings that RUNX1, known to be crucial for URE-PrPr interaction, occupies both the URE and the PrPr of the PU.1 locus provide a molecular understanding of locus-specific activation. We propose that once the URE and the PrPr are brought into close proximity, RUNX1 molecules that are parts of separate URE- and PrPr-bound complexes might interact, resulting in the formation of the URE-PrPr (enhancer-promoter) transcriptional activation complex. In supporting this mechanism, RUNX1 sites at enhancers and promoters have been shown to be critical for induction of CSF2 (encoding GM-CSF), CD34, and CEBPA (encoding C/EBPα),45-48 suggesting that RUNX1 could also contribute to specific enhancer-promoter docking at these gene loci. In line with this notion, locus-specific enhancer-promoter interaction could be induced by artificially tethering a transcription factor to a promoter.49 Our findings, therefore, support a model in which specific and on-target enhancer-promoter interactions are achieved by transcription factors, bound to specific motifs both at the enhancer and the target promoter, which are able to dimerize or multimerize, thereby helping to fuse enhancer and promoter transcriptional complexes together.
How chromatin-bound protein complexes at enhancers and target promoters are brought together in a highly specific manner is still poorly understood. Our findings offer several exciting avenues that might explain how locus-specific induction is accomplished. First, we demonstrated that LOUP modulates recruitment of RUNX1 to its binding motifs at both the URE and the PrPr, suggesting that LOUP might serve as an “RNA bridge,” bringing the separate RUNX1-containing URE and PrPr transcriptional complexes into proximity, which finally fused into an URE-PrPr complex via RUNX1 dimerization. Second, locus specificity might also be enhanced based on our finding that LOUP arises from the URE and acts in cis to modulate chromatin looping at the nearby PU.1 locus. Accordingly, even when a small number of transcripts are being produced, local molecular concentration of LOUP could be enriched enough to profoundly influence rapid PU.1 mRNA induction. Indeed, we found that LOUP is a low-abundance lncRNA but is enriched in the chromatin fraction. Third, we revealed that LOUP is expressed exclusively in myeloid cells. This could explain why RUNX1, which is expressed in diverse cell types, induces URE-PrPr interaction and PU.1 expression specifically in myeloid cells. These findings, together, provide mechanistic understanding of gene-specific enhancer-promoter interaction and cell type-specific gene induction.
Our findings also contribute to the growing body of knowledge with regard to molecular functions of lncRNAs. Indeed, among thousands of lncRNAs that arise throughout the genomes, only a few have been precisely mapped and molecularly characterized.23 The herein-described lncRNA LOUP, presenting as spliced and polyadenylated transcripts, binds the Runt domain of RUNX1 via a repetitive region. Whether the minor transcript, originating from a splicing event within the second exon of LOUP, exhibits other molecular functions remains an open question and is being investigated. To our knowledge, LOUP is the first cellular RNA-interacting partner of RUNX1 being reported. Remarkably, we also discovered that LOUP is downregulated by RUNX1-ETO. It is also worth mentioning that a normal allele of RUNX1 is retained alongside RUNX1-ETO fusion gene in t(8;21) AML cells,50 and that RUNX1-ETO is implicated in exerting opposing effects by competing with RUNX1 for binding to protein partners and to RUNX1-binding sites, but not affecting URE-PrPr interaction.42,51,52 Collectively, our findings uncover heretofore-unknown cross-regulation and molecular interactions of lncRNAs with transcription factors and their oncogenic derivatives, providing mechanistic understanding underlying their molecular functions.
In summary, we identified lncRNA LOUP with several important molecular features, including cell type–specific expression and harboring a RUNX1-binding platform enabling LOUP to coordinate with RUNX1 to drive long-range transcription of PU.1 in myeloid cells. LOUP, a downstream inhibitory target of the oncogenic fusion protein RUNX1-ETO, is capable of inducing myeloid differentiation and inhibiting cell growth. Our finding raises the possibility that RNA regulators of transcription factors represent alternative targets for therapeutic development and provide a molecular mechanism explaining, at least in part, how ubiquitous transcription factors contribute to enhancer-promoter communication in both a cell type- and a gene-specific manner and how their chimeric derivatives disrupt this normal regulation in leukemia.
Acknowledgments
B.Q.T. thanks Linus Tsai and Touati Benoukraf for technical advice. The authors thank the Iannis Aifantis Laboratory for the generous gift of the sgRNA cloning vector, Robert Welner, To-Ha Thai, and Dong-Er Zhang for insightful comments. The authors also thank Junyan Zhang, Li Ying, Zen Xi, Sophie Kellaway, Qiling Zhou, Georges Lacaud, and Constanze Bonnifer for assistance and helpful suggestions.
This work was supported by the following grants and awards: National Institutes of Health (NIH) National Cancer Institute grants K01 CA222707 (B.Q.T.), R50 CA211304 (A.K.E.), and R00 CA188595 (A.D.R.); an Italian Association for Cancer Research (AIRC) award (A.D.R.); a US Department of Defense (DOD) grant W81XWH-20-1-0518 as well as AIRC awards 5x1000 (“Metastatic disease: the key unmet need in oncology,” to the MYNERVA Project), #21267 (M.T.V.); NIH National Heart, Lung, and Blood Institute grant P01HL095489 (L.C.); and an award from the Xiu Research Fund (L.C.). This work was also supported by NIH grants R35 CA197697 (from the National Cancer Institute) and P01HL131477 (from the National Heart, Lung, and Blood Institute), the Singapore Ministry of Health’s National Medical Research Council under its Singapore Translational Research (STaR) Investigator Award, the National Research Foundation Singapore, and the Singapore Ministry of Education under its Research Centres of Excellence initiative (D.G.T.).
Authorship
Contribution: B.Q.T. and D.G.T. designed the study with contribution from A.K.E., P.B.S., M.T.V., P.P.P., L.C., S.S.K., and A.D.R.; B.Q.T., S.U., Y.Z., A.K.E., S.C., P.Z., H.Z., E.L., F.M., E.v.d.K., E.F., C.G., and A.R.T. performed experiments; B.Q.T., V.E.A., and G.H. analyzed ChIP-seq data; B.Q.T. and M.A.B. analyzed RIP-seq and scRNA-seq data; B.Q.T. and T.M.N. analyzed bulk RNA-seq data and performed CRISPRa; R.C. and D.E.T. designed and performed RIP-seq experiments; H.Y. and B.Q.T. analyzed TCGA data; C.-S.W. and B.Q.T. performed PhyloCSF analysis; B.Q.T. and S.U. drew schematics; B.Q.T. and D.G.T. wrote the manuscript with input from authors, especially A.K.E., E.L., M.A.B., T.M.N., P.B.S., M.T.V., P.P.P., and A.D.R.; and D.G.T. supervised the project.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Daniel G. Tenen, Cancer Science Institute of Singapore, #12-01, MD6, 14 Medical Dr, 117599 Singapore; e-mail: daniel.tenen@nus.edu.sg; and Bon Q. Trinh, Harvard Medical School Innitiative for RNA Medicines, Center for Life Sciences, 3 Blackfan Circle, Boston, MA 02215; e-mail: btrinh@bidmc.harvard.edu.
The sequencing data reported in this article have been deposited in the Gene Expression Omnibus database (accession number GSE140459 [https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE140459; enter token evstyyqkznmvtox]).
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked ”advertisement“ in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal