Key Points
Negative flow cytometric MRD after 1 course of induction therapy did not identify a favorable risk group of patients with ML-DS for whom HD-AraC could be eliminated.
Complex cytogenetics were associated with increased risk of relapse in MRD− patients after EOI-1 and not treated with HD-AraC.

Abstract
Myeloid leukemia in children with Down syndrome (ML-DS) is associated with young age and somatic GATA1 mutations. Because of high event-free survival (EFS) and hypersensitivity of the leukemic blasts to chemotherapy, the prior Children’s Oncology Group protocol ML-DS protocol (AAML0431) reduced overall treatment intensity but lacking risk stratification, retained the high-dose cytarabine course (HD-AraC), which was highly associated with infectious morbidity. Despite high EFS of ML-DS, survival for those who relapse is rare. AAML1531 introduced therapeutic risk stratification based on the previously identified prognostic factor, measurable residual disease (MRD) at the end of the first induction course. Standard risk (SR) patients were identified by negative MRD using flow cytometry (<0.05%) and did not receive the historically administered HD-AraC course. Interim analysis of 114 SR patients revealed a 2-year EFS of 85.6% (95% confidence interval [CI], 75.7-95.5), which was significantly lower than for MRD− patients treated with HD-AraC on AAML0431 (P = .0002). Overall survival at 2 years was 91.0% (95% CI, 83.8-95.0). Twelve SR patients relapsed, mostly within 1 year from study entry and had a 1-year OS of 16.7% (95% CI, 2.7-41.3). Complex karyotypes were more frequent in SR patients who relapsed compared with those who did not (36% vs 9%; P = .0248). MRD by error-corrected sequencing of GATA1 mutations was piloted in 18 SR patients and detectable in 60% who relapsed vs 23% who did not (P = .2682). Patients with SR ML-DS had worse outcomes without HD-AraC after risk classification based on flow cytometric MRD.
Introduction
Myeloid leukemia associated with Down syndrome (ML-DS)1 is a clinically and biologically distinct form of leukemia occurring in young children with Down syndrome (<4 years of age). ML-DS is characterized by a predominant megakaryoblastic phenotype, high prevalence of antecedent cytopenias,2 and somatic GATA1 mutations.3 Approximately 10% to 30% of newborns with DS first develop a preleukemic disorder termed transient abnormal myelopoiesis (TAM),1 previously also transient myeloproliferative disorder or transient leukemia.4,5 In ∼20% of patients with TAM, a subclone evolves into ML-DS, as evidenced by concordant clonal GATA1 mutations.6-9 Treatment of ML-DS has been associated with superior event-free survival (EFS) compared with acute myeloid leukemia (AML) in children without DS10-15 likely because of the hypersensitivity of ML-DS blasts to chemotherapy, including cytarabine,16,17 an agent historically used at high doses for the treatment of AML.
A series of clinical trials conducted by the Children’s Oncology Group (COG) and other study groups have aimed to maintain a high EFS while reducing intensity and toxicity of treatment, such as the cardiotoxicity associated with anthracyclines.18 In contrast, despite the increased drug sensitivity of ML-DS blasts, including to cytarabine, the use of high-dose cytarabine (HD-AraC) has been preserved in most treatment regimens for ML-DS, including the predecessor study, AAML0431, in which early introduction of HD-AraC was associated with an overall survival (OS) of 93.5% but the HD-AraC course was also associated with the majority of the observed infectious toxicity.14 Excellent survival outcomes combined with blast hypersensitivity to chemotherapy, an association of treatment-related morbidity with HD-AraC, and successful treatment of ML-DS without HD-AraC by another study group,12 prompted us to investigate whether elimination of HD-AraC was feasible in a favorable or standard risk (SR) group defined by negative measurable residual disease (MRD) by multidimensional flow cytometry after the first course of therapy, as suggested by AAML0431.14
By contrast, 10% to 15% of patients with ML-DS do not survive, primarily because of relapse or refractory disease,15,19-21 and in AAML0431, were more frequently identified by the presence of MRD after the first course of therapy.14 This high-risk group may benefit from intensification of upfront treatment.
Our study therefore introduced treatment stratification of chemotherapy for ML-DS using MRD measured by difference from normal (ΔN) flow cytometry at the end of the first course of induction (EOI-1) therapy to allocate patients to either a SR (MRD−) or high-risk (HR) (MRD+) arm. Treatment of SR patients eliminated the single course of HD-AraC, but otherwise mirrored AAML0431. In contrast, HR patients receive intensified chemotherapy modeled on that administered for high-risk AML in children without DS. Here, we report survival outcomes for the SR patients and examine the impact of cytogenetics, molecular MRD based on error-corrected sequencing (ECS) of patient-specific GATA1 mutations, and frequency of infectious complications.
Patients and methods
Trial design
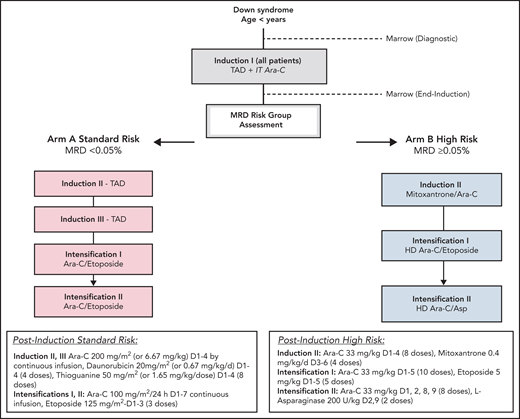
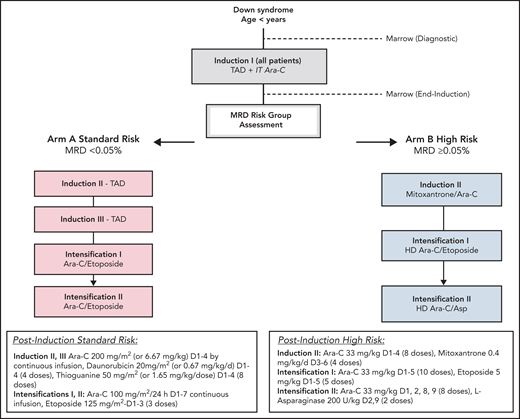
Eligibility criteria were as follows: children with DS (constitutional trisomy 21 or trisomy 21 mosaicism) and age >90 days and <4 years with (1) AML (≥20% bone marrow blasts) or (2) myelodysplastic syndrome (according to World Health Organization myelodysplastic neoplasm criteria22), or (3) patients more than 8 weeks since resolution of TAM with ≥5% blasts in the peripheral blood or increasing blast count (≥5%) in serial bone marrow aspirates at least 4 weeks apart. All patients received the same first course induction I (thioguanine 50 mg/m2/dose twice daily, days 1-4; cytarabine 200 mg/m2 per 24 hours' continuous infusion, days 1-4; daunorubicin 20 mg/m2 on days 1-4 over 1-15 minutes) and a single dose of age-based intrathecal cytarabine. For patients younger than 36 months, dosing was based on weight. After count recovery, residual disease in the bone marrow was measured by multidimensional flow cytometry. Patients with an MRD level below 0.05% were assigned to the SR arm (arm A). SR therapy consisted of 2 more courses of thioguanine, cytarabine, and daunorubicin followed by 2 identical courses of intensification therapy (intensification I and II: cytarabine 100 mg/m2 per 24 hours' continuous infusion, days 1-7; etoposide 125 mg/m2 per day, days 1-3). Patients with an MRD level of 0.05% or greater were assigned to the HR arm (arm B). Patients were not required to meet minimal organ function requirements before enrollment (Figure 1; supplemental Figure 1, available on the Blood Web site). The trial was approved by the Central Institutional Review Board of the National Cancer Institute and institutional review boards of each enrolling center. Patients and their families provided informed consent or assent as appropriate. The trial was conducted in accordance with the Declaration of Helsinki (NCT02521493).
Cytogenetics
Analyses were performed by COG-approved local and reference cytogenetic laboratories in accordance with the American College of Medical Genetics and Genomics and College of American Pathologists guidelines.21 A detailed summary of results designated according to the International System for Human Cytogenetic Nomenclature22 and images of representative karyograms and ancillary fluorescence in situ hybridization images were submitted by the laboratories for central review by 2 cytogenetics reviewers (S.R. and B.H.). Unbalanced abnormalities (those resulting in whole chromosome or partial chromosome gains and/or losses) were coded as follows: chromosome involved, short arm or long arm, gain or loss. The complete karyotype International System for Human Cytogenetic Nomenclature designation was retained to enable analysis for recurring rearrangements or breakpoints and for coding of balanced rearrangements. Each karyotype was scored for complexity defined as presence of 3 or more independent abnormalities, including ≥ structural abnormalities.23
Flow cytometry
Bone marrow samples were submitted at the end of the first course of induction therapy to a single central reference laboratory (Hematologics Inc, Seattle, WA) and stained with a standardized panel of monoclonal antibodies (supplemental Data), designed to detect residual disease by using the difference from normal approach (ΔN).24-29 Specimens were processed as previously described.24,25 Our analysis of ML-DS samples paid particular attention to avoiding misclassification of nonleukemic myeloid progenitors coexpressing CD56 and CD34, as well as CD33 (in addition to CD117, CD13, CD34, and CD45), which uniquely occur in regenerating normal bone marrow of children with DS after chemotherapy for AML or acute lymphoblastic leukemia,30,31 as residual leukemic disease. All data were reviewed by 2 independent analysts, who were blinded to patient information and came to agreement on a patient’s residual disease status before issuing a report. A level of 0.05% of total nucleated cells was chosen as the clinical cutoff based on threshold of detection and regulatory agency requirements. Results were reported through a Web-based platform (Rave EDC, Imedidata).
Statistical analysis
Data were current as of 31 March 2020, with a median follow-up of 2.1 (range, 0.48-3.6) years for SR patients. The significance of observed proportions was tested using Pearson’s χ2 and Fisher’s exact test when data were sparse. The Kaplan-Meier method was used to estimate OS and EFS. OS was defined as time from study entry, EOI-1, or from relapse until death. EFS was defined as time from EOI-1 to failure by not achieving complete remission, relapse, second malignancy, or death. Patients who experienced refractory disease with ≥5% bone marrow blasts after induction II or who experienced a relapse or were not in complete remission by the end of induction III were defined as an induction failure. The cumulative incidence of relapse was obtained by methods that account for competing events and was defined as the time from EOI-1 to relapse where deaths without a relapse were competing events. Gray’s test was used for comparisons. Patients lost to follow-up were censored at the date of last contact. Survival probabilities were reported with 95% confidence intervals (CI) calculated by the log–log transformation. This study was designed to compare SR patients against a fixed 2-year EFS of 93.5%, which was observed for comparable patients with ML-DS (ie, MRD− at end of induction I) treated on AAML0431. Assuming a null EFS of 93.5% at 2 years, there was 95% power to detect an alternative EFS of 87% at 2 years with 1-sided testing at the 10% level of statistical significance if there were 200 SR patients who continued to induction II. The study design included interim monitoring after ∼50% of the expected number of EFS events had been observed. Monitoring for insufficient EFS from the EOI-1 of the treatment of SR patients used monitoring based on the Lan-DeMets criterion with an α-spending function αt2 (truncated at 3 standard deviations) and 10% type I error. A Woolson 1-sample log-rank test was used to compare the observed EFS with AAML0431 EFS for MRD− patients who continued to induction II. EFS for AAML0431 was characterized by a separate cure model for SR patients: S(t) = 0.905 + (0.095) × exp(−0.00127 × t) where t was measured in days. A P value less than the boundary value of .025 for 50% information time would result in rejection of the hypothesis. The study specified comparing the Kaplan-Meier estimate of 2-year EFS (KM2) vs AAML0431 using a test statistic of In(−In(KM2)) − In(−In(0.935)))/(estimated standard deviation of KM2 × (In(−In(KM2))) compared with a standard normal distribution.
Results
Patient characteristics and accrual
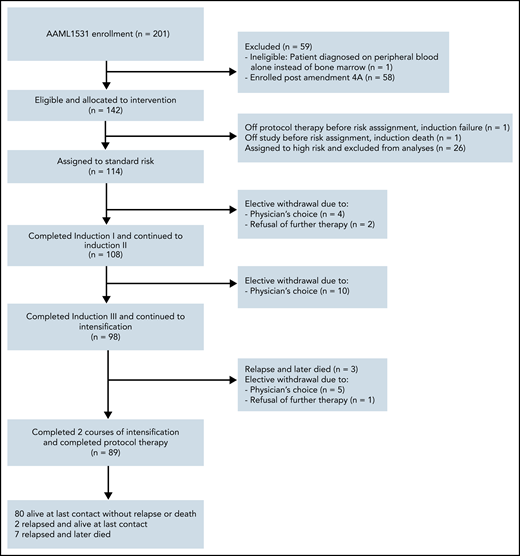
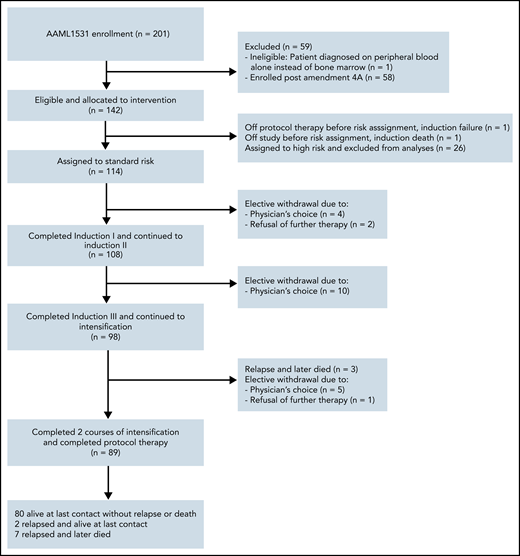
A total of 201 patients with DS (constitutional trisomy 21 or trisomy 21 mosaicism) were enrolled between November 2015 and March 2020 (Figure 2). One patient was ineligible because of the patient being diagnosed on peripheral blood alone instead of bone marrow. The study was amended on 30 October 2018 and the SR arm was closed to accrual. Fifty-eight patients were enrolled postamendment and are not included in this report. As for the remaining 142 patients, 2 patients were not assigned a risk group because of death during induction I or removal from study by physician’s choice. The remaining patients were assigned HR (n = 26) and SR (n = 114) by the EOI-1. Patient characteristics of those assigned to SR are shown in Tables 1 and 2 and supplemental Table 1.
Survival outcomes
An interim analysis, specified by the study protocol after half the expected EFS events had occurred, was performed in October 2018, when a total of 114 subjects had been enrolled on the SR arm (arm A). The observed 2-year EFS for SR patients (treated without HD-AraC) was 85.6% (95% CI, 75.7-95.5) compared with 93.5% (95% CI, 87.5-96.7) for patients who were MRD− at the end of induction course 1 on AAML0431 (which had included HD-AraC as the second course of overall therapy) (Figure 3A). This difference was statistically significant (P = .0002), met protocol criteria of inefficacy for AAML1531 SR therapy and led to closure of arm A. At 2 years, the OS was 91.0% (95% CI, 83.8-95.0) (Figure 3B). Twelve SR patients experienced a relapse, of which 11 occurred in the bone marrow and 1 was isolated to the central nervous system (CNS). All but 1 relapse event occurred within the first year from study entry (median time-to-relapse 208 days; range 136-511 days) (supplemental Table 2). The cumulative incidence of relapse for SR patients was 10.8% (95% CI, 5.9-17.4) at 2 years (supplemental Figure 1). For SR patients with relapse, OS was 16.7% (95% CI, 2.7-41.3) at 1 year (supplemental Figure 3). Of 12 SR patients who relapsed, two-thirds received relapse therapy with curative intent and 1 patient remains alive, including 1 who underwent allogeneic stem cell transplantation (supplemental Table 2).

Survival of standard risk (SR) patients with ML-DS on AAML1531. (A) The observed 2-year EFS for SR patients (treated without HD-AraC) was 85.6% (95% CI, 75.7-95.5) on AAAML1531 compared with 93.5% for patients who were MRD− at the end of induction course I but whose treatment on study AAML0431 had included HD-AraC. (B) 2-year OS (95% CI) for SR patients on AAML1531 was 91.0% (83.8-95.0) compared with 96.8% (91.6-98.8) for MRD− patients on AAML0431.
Survival of standard risk (SR) patients with ML-DS on AAML1531. (A) The observed 2-year EFS for SR patients (treated without HD-AraC) was 85.6% (95% CI, 75.7-95.5) on AAAML1531 compared with 93.5% for patients who were MRD− at the end of induction course I but whose treatment on study AAML0431 had included HD-AraC. (B) 2-year OS (95% CI) for SR patients on AAML1531 was 91.0% (83.8-95.0) compared with 96.8% (91.6-98.8) for MRD− patients on AAML0431.
Cytogenetics
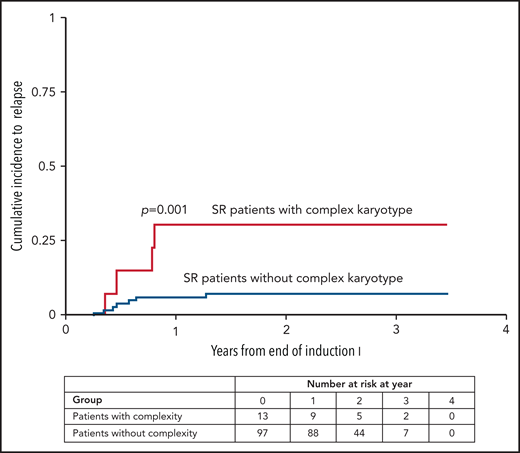
Of 110 SR patients with adequate cytogenetics, 91 (83%) had leukemic blasts with acquired clonal abnormalities in addition to constitutional trisomy 21. Most frequent were whole chromosome gains and structural rearrangements, leading to partial chromosome losses and gains. Among whole chromosome gains, trisomy 8 was most common (30% of patients), followed by gain of a fourth copy of chromosome 21 (16% of patients). Structural abnormalities present in at least 10% of patients included loss of 7p (13%) and gain of 1q (11%). Other recurring abnormalities present in at least 5% of patients included monosomy 7, trisomy 14, trisomy 11 or gain of 11q, and losses of 3q, 5p, or 13q. None of the recurring balanced rearrangements common to de novo non-DS AML were detected, and no novel recurring structural rearrangement was identified. A complex karyotype was present in 12% (n = 13) of patients with SR ML-DS. Complex karyotypes were significantly more frequent among SR patients who relapsed (n = 4/11) compared with those who did not (n = 9/99): 36% vs 9% (P = .0248). The cumulative incidence of relapse was 30.8% (95% CI, 8.8-56.5) at 2 years for SR patients with a complex karyotype compared with 7.5% (95% CI, 3.3-14.0) for those without (P = .001) (Figure 4).

Cumulative incidence of relapse according to complex karyotype. The cumulative incidence of relapse (95% CI) for AAML1531 SR patients with a complex vs non-complex karyotype was 30.8% (8.8% to 56.5%) vs 7.5% (3.3% to 14.0%) at 2 years.
Cumulative incidence of relapse according to complex karyotype. The cumulative incidence of relapse (95% CI) for AAML1531 SR patients with a complex vs non-complex karyotype was 30.8% (8.8% to 56.5%) vs 7.5% (3.3% to 14.0%) at 2 years.
MRD level by flow cytometry and ML-DS immunophenotype
EOI-1 bone marrow samples were available for all 114 SR patients and a diagnostic sample was available for comparison in 110 cases. A repeat sample was required for 3 patients after blood counts had further recovered because the EOI-1 bone marrow specimen was of inadequate quality. A post hoc re-analysis of EOI-1 MRD levels in the 12 SR patients who relapsed confirmed the negative MRD result (undetectable [ie, <0.02%]). One SR patient had an EOI-1 MRD value of 0.03%. This patient has not developed a relapse. See supplemental Data (including supplemental Figure 4 and supplemental Table 3) for a summary of data on MRD measurement by ECS of GATA1 mutations and targeted sequencing identifying additional mutations in ML-DS blasts, which were collected as part of an optional study component from a subset of 18 patients.
Toxicity of treatment
Time to ANC recovery, rates of febrile neutropenia, and intensive care unit (ICU) admissions per course of SR chemotherapy are shown in supplemental Table 4. Comparing course-to-course, despite identical chemotherapy, the proportion of patients with bacterial infection at sterile sites was lower than in AAML0431 (supplemental Table 5). There were no differences in rates of febrile neutropenia or ICU admissions in a course-to-course comparison excluding the HD-AraC course of AAML0431 (data not shown). Specific bacterial and fungal organisms are listed in supplemental Table 6. Of the 11 deaths on study, only 1 was due to toxicity. This patient experienced human metapneumovirus (hMPV) infection before and during induction I and died of respiratory failure. Microbiologically documented nonsterile site infections including viral infections are detailed in supplemental Table 7. During induction I, 6 patients experienced grade 3 or higher respiratory infection, including the patient who died of respiratory failure from hMPV. A second patient acquired hMPV during induction I and survived after a prolonged course of mechanical ventilation.
Similar to the predecessor study, SR patients received a cumulative anthracycline exposure of 240 mg/m2 doxorubicin equivalents. Because of bolus administration of anthracyclines in this study, use of dexrazoxane was feasible and permitted at the discretion of treating physicians and documented for 60% of patients in induction I, 58% in induction 2, and 59% in induction 3. Ejection fraction (EF) by echocardiogram was measured before each anthracycline-containing chemotherapy course and annually following completion of protocol therapy. No clinically detected cardiac dysfunction of any grade was reported. Subclinical cardiac toxicity was observed in only 3 patients: 1 with a grade 3 decrease in EF following induction I (from 65% to 32%) that resolved spontaneously; a second patient with a baseline reduced EF (38%), which normalized during therapy and declined after 3 years of follow-up (48%); and a third patient with a first reduction of EF (to 48% and 55%) after 1 and 2 years of follow-up, respectively. All 3 patients had received dexrazoxane with all anthracycline-containing treatment blocks. There was no association between use of dexrazoxane and relapse.
Discussion
Increased sensitivity of ML-DS blasts to chemotherapy drugs, including but not limited to cytarabine, and the desire to avoid excessive infectious and cardiac toxicity18 have led study groups to develop specific treatment protocols for ML-DS with decreasing treatment intensity.14,15,32 Most of these chemotherapy protocols, however, continue to incorporate courses of HD-AraC and none incorporates a stratification of treatment intensity according to relapse risk, in contrast to the majority of contemporary treatment approaches for childhood cancer. Prior to this COG study, the Japanese Childhood AML Cooperative Study Group observed a comparable 3-year EFS and OS after treatment of ML-DS without HD-AraC.12 The predecessor study AAML0431 showed that flow cytometric MRD at EOI-1 could significantly classify risk groups,14 but there remained persistently poor outcomes for the ∼10% to 15% of patients with relapsed or refractory ML-DS.14 Our study was designed to introduce risk stratification of treatment intensity for ML-DS while continuing on the trajectory of a therapy reduction for the majority of patients. SR patients were defined as MRD-negative (<0.05%) by multidimensional flow cytometry at EOI-1 and did not receive HD-AraC, whereas HR patients were directed to more intense consolidation (this arm continues to accrue and is not reported here). As a result, for the SR patients, the cumulative dose of cytarabine decreased from 27.8 g/m2 over 6 courses in AAML0431 to 3.8 g/m2 over 5 courses in the current study (supplemental Table 8). Because of the extreme rarity of CNS involvement by ML-DS, no patient was identified among 375 enrolled in 2 large recent studies,14,15 the number of intrathecal chemotherapy doses was further reduced from 2 to 1. Omission of HD-AraC, however, resulted in a statistically and clinically significant decrease of the 2-year EFS from 93.5% to 85.6%. Only 2 of 12 SR patients who relapsed survived, despite intensive relapse chemotherapy and stem cell transplantation, resulting in a dismal 16% OS. In contrast to others,20 we did not observe responses to relapse chemotherapy in patients who had previously not been treated with HD-AraC. Although flow cytometric MRD had identified a subgroup with extremely favorable prognosis (2-year EFS 93%) when treated with HD-AraC, negative MRD in our study did not identify a favorable risk group for whom HD-AraC was dispensable. We conclude that HD-AraC is an essential component of treatment to maintain the previously shown excellent EFS when using solely MRD by flow cytometry for therapy stratification.
Absence of any detectable EOI-1 MRD in the bone marrow of all 12 SR patients who relapsed suggests that this outcome is not explained by the slightly higher threshold for positivity used in the current study (0.05% vs 0.01% in AAML0431). In contrast to the prognostic value of EOI-1 MRD in non-DS AML,25 we conclude that flow cytometric EOI-1 MRD does not adequately identify patients with ML-DS who can safely benefit from a reduction in therapy intensity. Our results obtained from a subset of patients detected GATA1 mutations in 89% of ML-DS patients at diagnosis and confirmed the experience of others that false-negative results may occur because of a low blast percentage in the bone marrow sample, deletions that encompass primer annealing sites, and rare mutations mapping outside exons 2 and 3.3 They also suggest that MRD measurement by GATA1 ECS MRD is feasible in most but not all patients with ML-DS. Further work is needed, including the definition of a prognostically relevant cutoff variant allele frequency, before conclusions regarding the clinical utility of this new tool are possible.
Cytogenetic data from 110 SR patients confirm that the cytogenetic landscape of ML-DS is distinct from non-DS AML. As expected, none of the common recurring rearrangements of non-DS pediatric AML were identified. The majority of abnormalities in ML-DS blasts were unbalanced, resulting in gain or loss of chromosomal material. Trisomy 8, tetrasomy 21, gain of 1q, loss of 7p, and gain of 11q were identified as the most frequent abnormalities similar to the predecessor study,21 highlighting the comparability of study populations. These abnormalities have also been observed by other groups.33-36 Gains of 8, 21, and/or 1q are not exclusive to ML-DS and instead represent frequent secondary abnormalities in diverse hematologic malignancies.37 The small number of SR patients with relapse precludes prognostic conclusions for individual abnormalities. Monosomy 7, which has been variably described as either prognostically unfavorable,32 similar to non-DS AML, or neutral38 in ML-DS, was not more frequent among SR patients who relapsed. Of note, although a complex karyotype may39 or may not be42 prognostic in pediatric non-DS AML, our data suggest that a complex karyotype is associated with a higher risk of relapse in MRD− patients with ML-DS who are treated without HD-AraC.
Reduction of treatment-related mortality2 and anthracycline-related cardiomyopathy18 remains a goal for patients with ML-DS. The BFM study group reported microbiologically documented infections in 30% of patients, the majority of which were gram-positive bacteria, and highlighted the importance of serious viral infections (infection-related mortality was 4.9% and all 3 deaths were due to viral infections40). Therefore, the driving force behind the elimination of HD-AraC from the treatment of SR patients was the association of this chemotherapy course with the highest rates of grade 3 and higher febrile neutropenia (29.7%), sterile-site bacterial infections (22.6%), and ICU admissions (7%).14 Despite identical chemotherapy during all remaining courses, we observed a significantly lower rate of sterile-site infections, likely because of improvement in supportive care such as administration of prophylactic antibiotics during periods of prolonged neutropenia, data that were not captured by our study. Like others,40 we observed severe viral infections, specifically 2 patients with hMPV pneumonia and respiratory failure during induction 1, one of which was fatal, prompting a recommendation to participating centers to prioritize recovery of patients with ML-DS from acute viral infection over early start of protocol chemotherapy. Following this recommendation, no further cases of virally induced respiratory failure were observed.
Although long-term follow up for these patients will be critical to monitor for a decrease in cardiac function over time, short-term cardiotoxicity of AAML1531 SR protocol therapy appears to be minimal after use of dexrazoxane in the majority of patients.
In summary, our study shows the current limit of a further reduction in treatment intensity for ML-DS, specifically, the need to continue the administration of HD-AraC. We found that the prognostic value of flow cytometric MRD in patients with ML-DS needs to be assessed with caution and that MRD via flow cytometry alone is insufficient to identify patients for whom treatment intensity can be reduced. Incorporation of cytogenetics and more sensitive MRD methodologies, as well as molecular disease mechanisms are likely to be required to improve risk stratification of ML-DS and guide the integration of molecularly targeted small molecules or immunotherapy into future trials to reduce the number of patients who relapse and to improve overall outcomes.
Acknowledgment
The authors thank Vani Shanker for her constructive review of the manuscript.
This work was supported by grants from the National Institutes of Health National Clinical Trials Network (NCTN) Operations Center, National Cancer Institute (U10CA180886), National Clinical Trials Network (NCTN) Statistics & Data Center (U10CA180899), and the St Baldrick’s Foundation. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Authorship
Contribution: J.N.B., J.H., A.H.-M., J.T., and A.G. were involved in the design and planning of the trial; J.H., J.N.B., S.V., M.O., N.H., and R.S. were involved in the writing of the manuscript; A.B., S.R., and B.H. performed the cytogenetic analysis; L.B. and M.L. performed the flow cytometric studies; S.T. and T.D. performed and analyzed the sequencing studies; K.C. performed the hematopathology review; E.A.K. was involved in the concept design and editing the manuscript; and R.G., Y.-C.W., and T.A. oversaw the data collection and statistical analysis.
Conflict-of-interest disclosure: J.N.B. is a member of the Scientific Advisory Board of Oxford Immune Algorithmics and receives an honorarium for this role. The remaining authors declare no competing financial interests.
Correspondence: Jason N. Berman, CHEO Research Institute, 401 Smyth Rd, Ottawa, ON K1H 8L1, Canada; e-mail: jberman@cheo.on.ca.
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.