


Abstract
Genome-wide analyses have revealed that long noncoding RNAs (lncRNAs) are not only passive transcription products, but also major regulators of genome structure and transcription. In particular, lncRNAs exert profound effects on various biological processes, such as chromatin structure, transcription, RNA stability and translation, and protein degradation and localization, that depend on their localization and interacting partners. Recent studies have revealed that thousands of lncRNAs are aberrantly expressed in various cancer types, and some are associated with malignant transformation. Despite extensive efforts, the diverse functions of lncRNAs and molecular mechanisms in which they act remain elusive. Many hematological disorders and malignancies primarily result from genetic alterations that lead to the dysregulation of gene regulatory networks required for cellular proliferation and differentiation. Consequently, a growing list of lncRNAs has been reported to be involved in the modulation of hematopoietic gene expression networks and hematopoietic stem and progenitor cell (HSPC) function. Dysregulation of some of these lncRNAs has been attributed to the pathogenesis of hematological malignancies. In this review, we summarize current advances and knowledge of lncRNAs in gene regulation, focusing on recent progress on the role of lncRNAs in CTCF/cohesin-mediated 3-dimensional genome organization and how such genome folding signals, in turn, regulate transcription, HSPC function, and transformation. This knowledge will provide mechanistic and translational insights into HSPC biology and myeloid malignancy pathophysiology.
Introduction
One of the most compelling findings of the genome era of research is that the genome is extensively transcribed from noncoding regions and that these noncoding transcripts possess potential regulatory function.1,2 Noncoding transcripts include long noncoding RNAs (lncRNAs), which are >200 nucleotides in length with very little or no protein coding ability, and other distinct classes of noncoding RNAs, including microRNAs, small nuclear RNAs, and small nucleolar RNAs. Depending on their locations, lncRNAs can function in the nucleus, nucleolus, and cytoplasm. This review focuses on the nuclear function of several newly emerged lncRNAs with respect to the CCCTC binding factor (CTCF)/cohesin-associated genomic structural and transcriptional regulation in hematopoiesis and leukemogenesis.
Many functions of lncRNAs have been discovered to involve diverse biological processes, such as imprinting, X-chromosome inactivation, apoptosis, cellular proliferation and differentiation, stem cell biology, and tumorigenesis/leukemogenesis.3-8 The role of lncRNAs in transcriptional regulation involves recruitment or decoy of epigenetic regulators, transcription factors (TFs), and accessory proteins. However, how and where lncRNAs exert these diverse functions, including with what they interact, have become very important questions and an important research area in order to gain mechanistic insights into this class of RNA molecules. Acute myeloid leukemia (AML) is a heterogeneous disease with genetic or epigenetic alterations that systematically alter gene regulatory networks required for hematopoietic stem and progenitor cell (HSPC) proliferation and differentiation. A growing list of lncRNAs that are involved in controlling hematopoietic gene expression networks has been discovered, and some are linked to the dysregulation of HSPC function in AML.9,10 In this review, we focus on molecular mechanisms through which lncRNAs systematically regulate hematopoietic transcription pathways via their specific roles in 3-dimensional (3D) hematopoietic/leukemic genome organization.
Overview of mechanisms by which lncRNAs regulate gene transcription
In the nucleus, lncRNAs can regulate gene transcription in cis and trans actions via physical interactions with target DNA and associated proteins. lncRNAs can regulate epigenetic landscapes, enhancer/promoter activity, and RNA polymerase II machinery by diverse mechanisms.11,12
Scaffold
Xist lncRNA-mediated X-chromosome inactivation is a well-known example of lncRNA-driven transcriptional silencing (Figure 1A), one of the critical mechanisms leading to the gene dosage compensation in female cells. As a key regulator of this process, Xist lncRNA, via the orchestration of 3D chromatin conformation, spreads and coats polycomb repressive complex 2 (PRC2) and resulting H3K27me3 modifications on the future inactive X-chromosome for stable silencing of the entire X-chromosome.5,13 It is interesting to note that deletion of Xist in the hematopoietic compartment of female mice impaired hematopoietic stem cell (HSC) function, and mutant females developed aggressive myeloproliferative neoplasm and myelodysplastic syndrome that resulted from a dysregulated myeloerythroid fate decision of HSCs.14 Notably, the X-linked master erythroid TF Gata-1 was upregulated in Xist−/− females, perhaps as a result of X-linked gene overdosage in mutant females.14 Consistent with Xist RNA–dependent PRC2 complex repressive function, recent studies using RNA-dependent chromatin immunoprecipitation sequencing analysis revealed that the PRC2 complex generally requires RNA binding to guide chromatin occupancy for spreading of the H3K27me3 mark and its gene-repressive effects.15 Thus, the role of the PRC2-mediated chromatin repressive complex may require diverse scaffold/architectural RNA species.
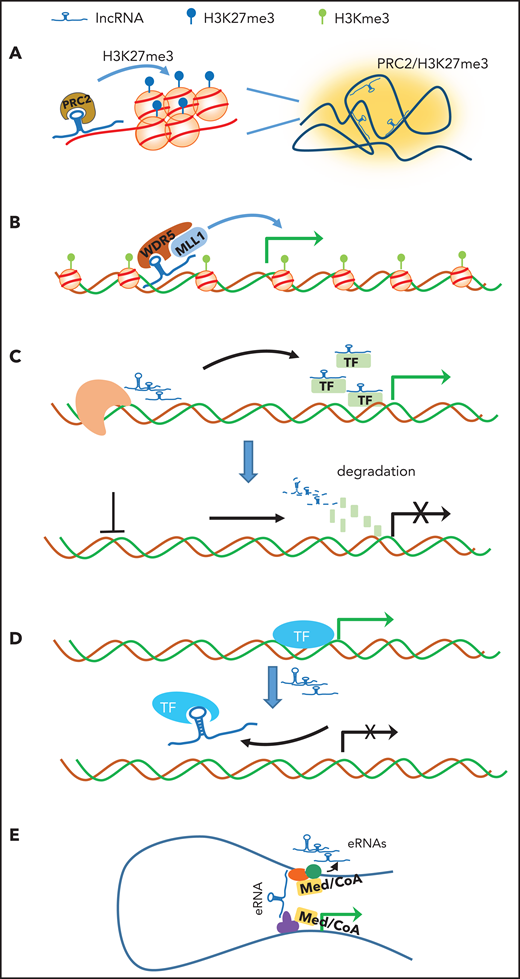
Schematic illustration of the potential mechanisms by which lncRNAs regulate chromatin structure and gene transcription. (A) Scaffold: lncRNA acts as a scaffold to orchestrate active or repressive chromatin (eg, PRC2 complex and H3K27me3 in Xi) over a large distance. (B) Recruitment/guide: lncRNA acts a mediator to recruit and guide the coactivator complex to enhancer/promoter for gene transcription. (C) Stabilization: lncRNAs produced in proximity of gene specifically associate with TFs or cofactors to facilitate gene transcription (upper panel). Depletion of lncRNA (dashed line) leads to degradation of TFs and cofactors, resulting in transcription silencing (lower panel). (D) Decoy: lncRNA acts as molecular decoy that dissociates promoter-bound TFs from the gene, leading to transcriptional inhibition. (E) Bridge: lncRNA or nearby enhancer-produced eRNA binds to and stabilizes the enhancer and promoter loop, leading to transcriptional activation. CoA, coactivator; Med, mediator.
Schematic illustration of the potential mechanisms by which lncRNAs regulate chromatin structure and gene transcription. (A) Scaffold: lncRNA acts as a scaffold to orchestrate active or repressive chromatin (eg, PRC2 complex and H3K27me3 in Xi) over a large distance. (B) Recruitment/guide: lncRNA acts a mediator to recruit and guide the coactivator complex to enhancer/promoter for gene transcription. (C) Stabilization: lncRNAs produced in proximity of gene specifically associate with TFs or cofactors to facilitate gene transcription (upper panel). Depletion of lncRNA (dashed line) leads to degradation of TFs and cofactors, resulting in transcription silencing (lower panel). (D) Decoy: lncRNA acts as molecular decoy that dissociates promoter-bound TFs from the gene, leading to transcriptional inhibition. (E) Bridge: lncRNA or nearby enhancer-produced eRNA binds to and stabilizes the enhancer and promoter loop, leading to transcriptional activation. CoA, coactivator; Med, mediator.
Recruitment/guide
Hox genes are critical for maintaining the balance between the self-renewal and differentiation of HSCs. Their dysregulation is a dominant mechanism underlying AML pathogenesis.16 Recently, it was reported that hundreds of lncRNAs are transcribed from HOX gene clusters.17 Many HOX genes can be regulated by specific lncRNAs via cis- or trans-action. One such lncRNA, HOTTIP, is transcribed from the distal HOXA locus and activates genes in cis by recruiting and guiding the MLL1/WDR5 complex to the posterior HOXA genes18 (Figure 1B); this suggests that lncRNAs can modulate chromatin dynamics by recruiting histone-modifying and chromatin-remodeling complexes to specific genomic loci. We recently discovered and cloned a unique HoxB locus–associated lncRNA, HoxBlinc, which is expressed during early hematopoietic differentiation and is strongly concomitant with H3K4me3 patterning and anterior Hoxb gene activation. HoxBlinc associates with the Setd1a/MLL1 histone methyltransferase complex to coordinate activation of the anterior Hoxb genes and specify hematopoietic cell fates during early hematopoietic differentiation.3 It is clear that lncRNAs can act as epigenetic regulators to modulate gene transcription.
Stabilization
Transcription regulation is central in controlling cellular proliferation and differentiation. Many proteins that are known to be involved in eukaryotic transcription can interact with DNA elements, as well as are involved in RNA interaction. A significant number of TFs can be modulated by specific lncRNAs, and their interaction may stabilize TF in chromatin template (Figure 1C). One example of such an lncRNA is PVT1, an lncRNA required by MYC in MYC-dependent cancers.19 Depletion of PVT1 led to disproportionately decreased MYC protein levels in MLL-AF9–driven AML, whereas enforced expression of Myc rescued the effects caused by Pvt1 knockdown.20 Threonine 58 phosphorylation enhances MYC protein degradation.21 The underlying molecular action of PVT1 in AML involves stabilization of MYC protein by inhibiting its T58 phosphorylation, thereby enhancing MYC-mediated transcription.
Decoy
lncRNAs can also inhibit the genomic binding of TFs by acting as a “decoy molecule.” The lncRNA growth arrest-specific 5 (Gas5) is involved in starvation-associated repression of the glucocorticoid receptor (GR)-mediated transcription.22GAS5 used a decoy mechanism to sequester GR from the glucocorticoid-responsive genes by the formation of an RNA motif in one of its stem-loop secondary structures that mimics the hormone response element–like DNA motif. Therefore, GAS5 competes for the binding of GR to the glucocorticoid-response elements in the promoters of glucocorticoid-responsive genes22 (Figure 1D).
Bridge
Enhancer RNAs (eRNAs) are a specific class of lncRNAs that are transcribed from enhancer DNA sequences and may have a profound impact on gene transcription. The p53-associated eRNAs were transcribed from the p53-bound enhancer regions (p53BERs) that induce p53-independent intrachromosomal interactions, bringing p53BERs into close proximity of the p53 target genes for efficient transcription enhancement of the associated p53 target genes and leading to p53-dependent cell cycle arrest23 (Figure 1E).
In T-cell acute lymphoblastic leukemia (T-ALL), mutation in the TAL1 gene is one of the most frequent gain-of-function mutations found in patients with T-ALL. The TAL1 transcription complex consists of multiple TFs, such as TAL1, GATA-3, RUNX1, MYC, and MYB, and acts as an oncogenic regulator circuit in TAL1 activation–driven T-ALL. One of the major targets of the TAL1 transcription complex is the proto-oncogene ARID5B, which supports the survival of human T-ALL cells and potential tumor formation in the thymus. TAL1 transcription complex binds to the −135 kb enhancer of the ARID5B gene and induces transcription of the eRNA ARIEL, which, in turn, stabilizes the enhancer-occupied TAL1 transcription complex and facilitates enhancer/promoter communications, leading to transcription activation of ARID5B and a positive feed-forward regulatory loop that promotes leukemogenesis.24
The RNA/DNA hybrid: R-loop or triple helix
As epigenetic and transcriptional regulators, lncRNAs need to access specific genomic loci, including enhancers and promoters, to exploit their regulatory effects. lncRNAs can use the R-loop or triple helix to access specific genomic regulatory elements and exert transcription regulation.4,25,26 R-loops are 3-stranded nucleic acid structures comprising RNA hybridized with the DNA template, leaving the nontemplate DNA single stranded. The presence of nascent RNA-dependent R-loops in the gene promoter region has been linked to transcriptional regulation through the modulation of promoter accessibility, RNA polymerase II recruitment, and RNA splicing.27-30 Recently, it was shown that APOLO lncRNA recognizes and coordinates expression of several target genes by the formation of R-loops in the Arabidopsis thaliana genome,25 suggesting that the R-loop underlies lncRNA-mediated genome accessibility for transcriptional regulation (Figure 2A). On the other hand, oncogenic lncRNA Khps1 can directly bind to the promoter of the proto-oncogene SPHK1 by formation of a DNA-RNA triplex through a noncanonical Hoogsteen base-pairing mechanism. Khps1 lncRNA then recruits and targets p300/CBP to the SPHK1 promoter, leading to local changes in chromatin structure and the activation of SPHK1 expression in an E2F1-dependent manner (Figure 2B).4 In the β-globin locus, an RNA transcript embedded in the second intron of the human β-globin gene forms a triplex with hypersensitive site 2 of the β-globin locus control region. The triplex structure displaces TFs and the RNA polymerase II complex, resulting in downregulation of genes controlled by hypersensitive site 2 in the locus.31 Thus, the direct binding of lncRNAs to enhancers and promoters may serve as a signal to initiate the transcription or silencing process.

LncRNA binds and accesses the regulator DNA elements by triplex or R-loop mechanism. (A) LncRNA forms a triple helix structure via a Hoogsteen base pair mechanism to access genomic DNA regulatory elements. (B) lncRNA forms an R-loop structure with the genomic DNA regulatory elements.
LncRNA binds and accesses the regulator DNA elements by triplex or R-loop mechanism. (A) LncRNA forms a triple helix structure via a Hoogsteen base pair mechanism to access genomic DNA regulatory elements. (B) lncRNA forms an R-loop structure with the genomic DNA regulatory elements.
Role of lncRNAs in shaping local chromatin structure and enhancer/promoter interactions
Genes and their regulatory elements reside nonrandomly in the nucleus of cells. A special genome organization involved in the positioning and organization of chromatin to facilitate the transcriptional machinery within the nucleus is essential for overall genome function. It seems that these topological associated chromatin domains (TADs) require structural/regulatory protein complexes, various genetic elements, and a nuclear envelope, as well as lncRNAs. Recent genomic studies revealed that many lncRNAs play an important role in local chromatin domain organization and enhancer/promoter interactions, and the list is still growing (Table 1). The Xist lncRNA–driven X-chromosome inactivation throughout the female life to equalize X-linked gene dosage with male is an excellent example of an lncRNA serving as a driving force for large-scale chromosome organization. Xist initiates and spreads PRC2 and the resulting H3K27me3 accumulation along the X-chromosome by orchestrating a 3D genome topology,5 as well as targets CTCF to specific genome loci to mediate long-range chromosomal interactions in a locus-specific manner during X-chromosome inactivation.32 Mechanistically, Xist lncRNA actively repels the cohesin complex from the inactive X-chromosome, because deletion of Xist restores cohesin binding. This results in reorganization of TADs that resemble an active X-chromosome,33 suggesting a central role for lncRNA in the 3D organization of the mammalian genome, perhaps mediated by CTCF and its associated cohesin complex.
lncRNAs involved in genome topology
| Name | Role in genome organization | Function | Reference |
|---|---|---|---|
| HOTTIP | Topological organization of posterior HOXA and canonical Wnt/β-catenin target genes | HSC self-renewal AML leukemogenesis | 7 |
| Xist | Scaffold and 3D topology for PRC2/H3K27me3 | X-inactivation | 33 |
| APOLO | R-loop–mediated decoy of PRC1 and chromatin loop modulation | Gene transcription root development | 25 |
| Firre | Spatial organization of multiple chromosomal regions | Adipogenesis ESC pluripotency | 67 |
| HOXBLINC | 3D organization of anterior HOXB genes | Hemagiogenic mesoderm NPM1C+-driven AML | 3, 44 |
| PAIR | PAIR elements/Eμ long-range interactions | VDJ recombination | 68 |
| MYMLR | Long-range enhancer/promoter interaction | MYC transcription | 69 |
| H19X | Long-range enhancer/promoter interaction | Transcription silencing | 70 |
| p53BER eRNA | p53BER and p53 target gene long-range contacts | p53-driven transcription | 23 |
| ARIEL eRNA | Long-range enhancer and promoter interaction | Proto-oncogene transcription T-ALL survival pathway | 24 |
| Name | Role in genome organization | Function | Reference |
|---|---|---|---|
| HOTTIP | Topological organization of posterior HOXA and canonical Wnt/β-catenin target genes | HSC self-renewal AML leukemogenesis | 7 |
| Xist | Scaffold and 3D topology for PRC2/H3K27me3 | X-inactivation | 33 |
| APOLO | R-loop–mediated decoy of PRC1 and chromatin loop modulation | Gene transcription root development | 25 |
| Firre | Spatial organization of multiple chromosomal regions | Adipogenesis ESC pluripotency | 67 |
| HOXBLINC | 3D organization of anterior HOXB genes | Hemagiogenic mesoderm NPM1C+-driven AML | 3, 44 |
| PAIR | PAIR elements/Eμ long-range interactions | VDJ recombination | 68 |
| MYMLR | Long-range enhancer/promoter interaction | MYC transcription | 69 |
| H19X | Long-range enhancer/promoter interaction | Transcription silencing | 70 |
| p53BER eRNA | p53BER and p53 target gene long-range contacts | p53-driven transcription | 23 |
| ARIEL eRNA | Long-range enhancer and promoter interaction | Proto-oncogene transcription T-ALL survival pathway | 24 |
ESC, embryonic stem cell; T-ALL, T-cell acute lymphoblastic leukemia.
The notion that lncRNAs are involved in CTCF-mediated genome organization stems from a seminal finding by Felsenfeld and colleagues that DEAD-box RNA helicase p68 and steroid receptor RNA activator (SRA) interact with CTCF and cohesin. Depletion of p68 or SRA impaired chromatin insulator activity by disrupting CTCF/cohesin interaction,34 suggesting that p68/SRA stabilizes the CTCF/cohesin complex and that RNA may be required for proper chromatin boundary function. Given that many lncRNAs are cis or trans acting35 and associated with CTCF,32 lncRNAs may contribute to the recruitment and stabilization of the CTCF complex in genomic loci. In support of this notion, 2 back-to-back publications in Molecular Cell demonstrated that the zinc finger (ZF) domains of CTCF are required for CTCF clustering and CTCF-mediated long-range chromatin interactions.36,37 The RNA-binding regions (RBRs) of CTCF are located within the ZF1 and ZF10 domains. RBRs are dispensable for DNA recognition and binding, but deletion of RBR disrupts half of the CTCF loops.37 Thus, it seems that CTCF contributes to genome topological regulation via an RNA-dependent mechanism. However, the type(s) of RNAs that contribute to CTCF genome action, how RNA species access specific genome regulatory loci, and more importantly, the identification of the underlying molecular mechanisms and biological consequences remain to be determined.
Role of lncRNAs in HSC biology
Compared with messenger RNA, the expression levels of lncRNAs are relatively low, yet the expression of many lncRNAs is highly cell-type/lineage specific, differentiation stage specific, or even disease state specific,38-40 suggesting that lncRNAs act as determinants of lineage commitment and differentiation, perhaps by acting coordinately with lineage-specific TFs. HSCs require a unique gene-regulatory program that reinforces their cellular identity and controls cell fate decisions. Recent comprehensive genome-wide studies on HSCs and lineage-primed multipotent progenitors revealed that hundreds of lncRNAs are expressed together with lineage-specific TFs in these cells.41,42 Many of these lncRNAs are closely associated with lineage-specific TFs that are required for hematopoietic differentiation and cell fate decision,41,42 suggesting that lncRNAs may provide another layer of regulation operating within the HSC hierarchy.
Consistently, Goodell and colleagues used extremely deep RNA sequencing (>1.3 billion combined HSC reads), which overcomes the relatively low expression levels of lncRNAs compared with protein coding transcripts in rare HSC populations, to identify lncRNAs that are specifically associated with mouse long-term HSCs.43 They identified and annotated 159 candidate HSC-specific lncRNAs with high confidence scores. Some of them modulate HSC self-renewal and differentiation by binding to hematopoietic TF binding sites.43 Among them, lncHSC-2 controls HSC self-renewal and differentiation by modulating the activity of basic helix-loop-helix TF, TCF3/E2A, through binding to the TCF3-bound E-box elements at genes that are important for HSC homeostasis. Loss of lncHSC-2 in HSPCs resulted in impaired HSC self-renewal and skewed T-cell differentiation by abrogating TCF3 binding, a TF that is required for HSC function and T-cell development.43 Thus, it is likely that lncRNAs act as an additional layer of genetic circuitry by modulating the accessibility and activity of TFs at their bound DNA motifs.
The expression of HOX genes, particularly HOXA and HOXB, is tightly regulated throughout hematopoiesis, with the highest expression in HSPCs that gradually decreases to minimum levels in mature lineages. Two HOX gene–associated lncRNAs, HOXBLINC and HOTTIP, were recently shown to play critical roles in HSC self-renewal and lineage commitments.3,7,44HOXBLINC, embedded in the anterior HOXB locus, is highly expressed in long- and short-term HSCs, and its expression diminishes in mature hematopoietic lineages.3,44 The expression pattern of HOXBLINC in hematopoiesis suggests a role in HSC maintenance and regulation. During embryonic development, induction of HoxBlinc transcription initiates anterior HoxB genes activation by recruiting the Setd1a/MLL1 complex to the promoters of anterior HoxB genes and promoting 3D enhancer/promoter interactions (Figure 3A). This regulatory process initiates and stimulates hemangiogenic Flk1+ mesoderm differentiation and the hematopoietic fate decisions.3HOXBLINC was aberrantly activated in patients with specific subtypes of AML carrying nucleophosmin 1 (NPM1C+) mutations. Interestingly, transgenic overexpression of HoxBlinc in the hematopoietic compartment results in aberrant HSC self-renewal and expanded myelopoiesis, leading to the development of AML-like disease that resembles NPM1C+-driven AML. Mechanistically, HOXBLINC acts as a downstream effector of NPM1C+ to recruit the MLL1 complex to the promoters of NPM1C+ signature genes and activate signature NPM1C+-associated transcription networks, including anterior HOXB genes.44
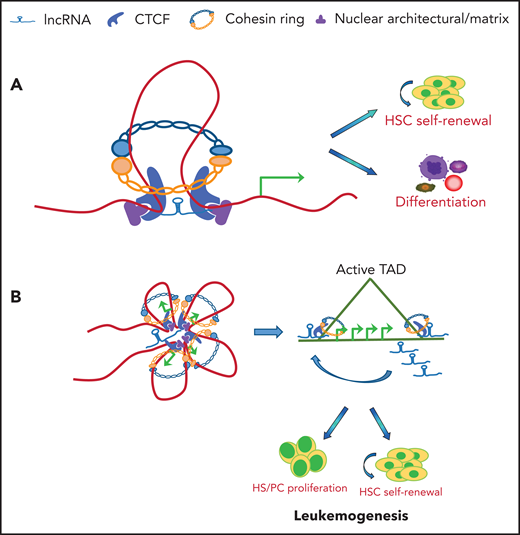
Schematic illustration of lncRNA involved in CTCF/cohesin-mediated chromatin interaction and genome topology. (A) CTCF and cohesin are frequently localized at enhancers and promoters in the genome. lncRNAs may tether and stabilize CTCF/cohesin-mediated enhancer/promoter communication by forming chromosomal loops, which lead to activation of genes required for HSC self-renewal or lineage commitment and differentiation. (B) lncRNA may coordinate with CTCF/cohesin complexes to form TAD domains in hematopoietic/AML genome. In both cases, chromatin organization results in systematic changes in gene regulatory networks and transcription programs that contribute to the promotion of hematopoietic lineage differentiation or blockage of lineage differentiation and leukemogenesis.
Schematic illustration of lncRNA involved in CTCF/cohesin-mediated chromatin interaction and genome topology. (A) CTCF and cohesin are frequently localized at enhancers and promoters in the genome. lncRNAs may tether and stabilize CTCF/cohesin-mediated enhancer/promoter communication by forming chromosomal loops, which lead to activation of genes required for HSC self-renewal or lineage commitment and differentiation. (B) lncRNA may coordinate with CTCF/cohesin complexes to form TAD domains in hematopoietic/AML genome. In both cases, chromatin organization results in systematic changes in gene regulatory networks and transcription programs that contribute to the promotion of hematopoietic lineage differentiation or blockage of lineage differentiation and leukemogenesis.
On the other hand, HOTTIP, which is transcribed from the posterior HOXA locus, activates multiple homeotic TF-encoded genes, including posterior HOXA genes, the canonical Wnt/β-catenin pathway, and various key hematopoietic regulators, by orchestrating CTCF-defined hematopoietic-associated TADs. Aberrant expression of HOTTIP in the mouse bone marrow hematopoietic compartment enhanced HSC self-renewal, leading to leukemic transformation of HSCs via aberrant binding of HOTTIP to hematopoietic/leukemic-specific TFs or CTCF motifs.7
In addition to those lncRNAs involved in HSC self-renewal and differentiation, maternal, but not paternal, deletion of H19 lncRNA and its associated upstream differentially methylated region (DMR) reduced adult HSC quiescence, which plays a key role in long-term HSC maintenance. The expression of H19 is maternally imprinted (expressed exclusively by the maternally inherited allele) and is regulated by binding of CTCF to the unmethylated DMR. Loss of maternally expressed H19 upregulated the paternally associated Igf2 gene pathway that resulted in FoxO3-mediated cell cycle arrest.6
Involvement of HOX-associated lncRNAs in AML leukemic genome organization
AML is a heterogeneous hematopoietic stem or progenitor cell disease that results from genetic alterations or somatic mutations in genes that are required for HSPC biology. Given that lncRNAs are crucial regulators of gene expression in HSCs that determine cell fate and commitment decisions, the expression landscapes of lncRNAs that exhibit an HSPC signature are likely shared with AML blasts.45 Many AML-associated lncRNAs are essential for the maintenance of leukemic transcription programs that promote leukemic stem cell signatures.20 Thus, the expression landscape of a specific lncRNA class may be of great prognostic value and clinical relevance for patients with AML.
NPM1 is the most commonly mutated gene and accounts for 30% of AML patients with a normal cytogenetic karyotype. NPM1C+ mutation results in aberrant cytoplasmic translocation of the encoded mutant nucleolar protein, which maintains a unique expression pattern of HOXA/B genes and the MEIS1 oncogene to facilitate AML leukemogenesis. An anterior HOXB-derived lncRNA, HOXBLINC, which is aberrantly activated in patients with AML carrying NPM1C+, mediates and maintains NPM1C+-derived leukemic gene signatures and leukemogenesis.44HoxBlinc activation is also required to promote HSPC transcription signatures and determine HSPC fate and commitment.3 In HSCs and AML, HOXBLINC RNA exploits an active 3D interactome within the anterior HOXB domain to recruit the MLL1/Setd1a complex and deposit H3K4me3 marks on each of the anterior HoxB genes for their activation.3,44 Given that NPM1 interacts with CTCF and provides nuclear matrix for anchoring CTCF-mediated chromatin loops,46 it would be interesting and important to determine how NPM1C+ alters hematopoietic genome topology and whether subsequent activation of HOXBLINC reorganizes and facilitates HSPC survival pathways upon cytoplasmic translocation of the NPM1C+ mutant.
The posterior HOXA locus–associated lncRNA, HOTTIP,18 exemplifies the importance of lncRNAs in leukemic genome organization. HOTTIP coordinates with CTCF to promote aberrant posterior HOXA TADs and chromatin signature in AML.7 Posterior HOXA genes are aberrantly activated in specific types of AML, especially in those patients and cells carrying mixed lineage leukemia (MLL) gene rearrangements (MLLr+) or NPM1C+ mutations.47,48HOXA and HOXB genes are critical for maintaining the balance between self-renewal and differentiation of HSCs.49-51 Dysregulation of HOXA and/or HOXB genes is a dominant mechanism of leukemic transformation.52 Epigenetic analysis of the HOX gene loci in MLLr+ or NPM1C+ AML cells revealed that distinct chromatin/epigenetic signatures were defined by CTCF boundaries in the posterior HOXA TAD domain, which contribute to aberrant HOXA gene expression in a HOTTIP-dependent manner.53,54 Attenuation of CBS7/9, a CTCF boundary located between the HOXA7 and HOXA9 genes in the beginning of the posterior HOXA TAD, alters posterior HOXA TADs and inhibits posterior HOXA gene signatures, including HOTTIP lncRNA, to alleviate AML leukemogenesis.53 Interestingly, depletion of HOTTIP also impairs the posterior HOXA TAD and oncogenic homeotic transcription program by impairing HOTTIP chromatin binding, leading to blockage of AML leukemogenesis.7 Overexpression of HOTTIP restores CTCF-mediated HOXA TADs and leukemogenesis in attenuated AML cells in the CBS7/9 boundary,7 supporting a feed-forward mechanism by which collaboration of HOTTIP and CTCF stratifies the CTCF boundary and enforces oncogenic posterior HOXA TADs and gene activation to promote aberrant HSPC self-renewal and transformation (Figure 3B). To support the collaboration between HOTTIP and CTCF in the TAD boundary, analyses of HOTTIP chromatin-binding motifs showed a significant enrichment of core CTCF consensus-binding motifs; loss of HOTTIP resulted in the decrease in a subset of CTCF-defined TADs and chromatin signatures in loci involved in homeotic oncogenic TFs and hematopoiesis.7 However, it remains unknown through which mechanisms lncRNAs, such as HOTTIP, are targeted to and/or recognize specific CBSs. It is also important to determine how widely required RNAs are for CTCF-mediated genome organization and the molecular basis by which lncRNAs act on CTCF-mediated topological genome regulation. Unbiased isolation of the HOTTIP-associated protein complex in AML cells would provide mechanistic insight into its action in CTCF-mediated genome organization.
lncRNAs and their pathways serve as biomarkers and potential therapeutic targets for leukemia
Many lncRNAs exhibit lineage-, differentiation stage-, or disease-specific expression, making them excellent candidates for biomarkers or therapeutic applications. A study evaluated the lncRNA expression profiles from 148 untreated older AML patients with normal cytogenetics to determine whether lncRNAs are associated with clinical features and certain recurrent mutations in these patients. Interestingly, distinct lncRNA expression profiles were highly associated with specific mutations, such as FLT3-ITD, NPM1, CEBPA, IDH2, ASXL1, and RUNX1 genes, whereas another subset of lncRNAs is highly correlated with treatment response and survival.55 Recently, RNA sequencing, followed by univariate and multivariate time-to-event analyses, of 274 Swedish patients with AML after intensive chemotherapy found that 33 lncRNAs were associated with overall survival, and their expression profiles predicted outcomes.56 These lncRNAs likely play oncogenic roles via their impact on 1 or several hallmarks of leukemia, including self-renewal, antiapoptosis, proliferation, and differentiation. The importance of some of these lncRNAs in reshaping the epigenetic landscape of the leukemic genome is beginning to be recognized. These AML-associated specific characteristics of lncRNAs make them potential markers for clinical prognostic outcomes and potential targets for treatment.
HOTTIP and HOXBLINC are HOX-associated oncogenic lncRNAs that are aberrantly activated in patients with AML carrying MLLr+ or NPM1C+ mutations.7,44HOTTIP lncRNA activation promotes HSC/leukemic stem cell (LSC) self-renewal and AML progression by activating HOXA9 and the canonical Wnt/β-catenin pathway,7 which is uniquely required for LSC self-renewal.57,58 Mechanistically, HOTTIP acts downstream of MLLr+ or NPM1C+ by coordinating TAD organization of the AML genome, perhaps by regulating CTCF chromatin boundary activity via direct binding to a subset of CTCF core motifs in the AML genome. Analyses of HOTTIP DNA binding motifs in the AML genome revealed a significant enrichment of CTCF core motifs,7 supporting that HOTTIP is involved in CTCF boundary function. Indeed, depletion of HOTTIP attenuates AML progression by impairing leukemic TADs and HOXA9- and/or β-catenin–driven transcription programs. In light of understanding the downstream pathways that mediate HOTTIP oncogenic action in MLLr+ AML, pharmacological targeting of the canonical Wnt pathway to suppress the β-catenin–mediated transcription program blocked MLLr+ LSC self-renewal and HOTTIP activation-driven leukemogenesis.7 Similarly, HOXBLINC activation in NPM1C+ AML cells specifically promotes the NPM1C+-driven transcription signatures to facilitate NPM1C+-derived AML leukemogenesis.44 Thus, small interfering RNAs/short hairpin RNAs or CRISPR/Cas9-targeted degradation or silencing of HOTTIP or HOXBLINC may enable systematic reversal of the genome TAD and transcription program driven by MLL fusions or NPM1C+. In addition, dead Cas9 targeted interference of interactions between lncRNAs and genetic elements or regulatory proteins at specific loci may be able to block specific leukemic transcription programs (eg, HOXA9 or β-catenin), leading to efficient targeted therapy
Summary and perspectives
Novel technologies have been used extensively in genome research to interrogate the genetic targets and associated protein complexes of specific lncRNAs in cells. As such, the roles of lncRNAs in lineage- or disease-specific genome organization have broadened in the past several years. However, the detailed mechanisms involved in lncRNA-dependent genome topology remain to be explored. Although HOTTIP lncRNA is shown to mediate TAD formation in several critical hematopoietic/leukemogenic loci during AML progression,7 many questions remain unanswered. Mechanistically, it remains unclear how lncRNA regulates TAD and TAD boundaries. It also remains to be determined whether and how lncRNAs recruit, bridge, guide, or facilitate the CTCF/cohesin complex to specific genomic regions to alter genome topological structure. CTCF is a master regulator of mammalian genome organization.59 CTCF modulates the chromatin TAD boundary, as well as enhancer/promoter contacts within TADs, in cohesin-dependent or -independent manners.60,61 Although the CTCF core motif is highly enriched in HOTTIP-bound genomic regions,7 it remains to be determined whether lncRNAs directly regulate the ability of CTCF to access chromatin DNA or its association with the cohesin complex. The cohesin complex frequently colocalizes with CTCF in the genome, and their binding sites are mutational hotspots in the noncoding cancer genome.62 In addition, the cohesin genes are frequently mutated in AML that exhibits a strong association with the NPM1 mutation.63 Therefore, it appears that the CTCF/cohesin-mediated function plays a critical role in HSPC regulation and AML leukemogenesis, perhaps through its actions in hematopoietic specific genome topology. A recent study showed that architectural RNA is essential for PRC2 complex chromatin occupancy,15 suggesting that RNA provides an additional regulatory layer and may have a wide role in chromatin domain organization beyond the PRC2-repressive complex. Given the general role of RNAs in chromatin and gene regulation, it will be interesting to determine whether the role of CTCF/cohesin requires general action from RNA, such as architectural RNA and regulatory lncRNAs in different AML subtypes, and whether such RNAs control CTCF DNA binding or CTCF/cohesin interaction. With new genomic techniques surging and increasing evidence of lncRNA’s role in AML genome organization, it will become clear whether CTCF/cohesin-mediated genome organization generally depends on RNAs to facilitate their activity in inducing aberrant genome topology in lineage- or leukemic-specific loci to promote leukemogenesis. More importantly, it remains to be determined whether lncRNAs play architectural or regulatory roles in CTCF/cohesin chromatin boundary activity and CTCF-mediated genome topology.
The advances in new RNA centric genomic/proteomic technologies, such as chromatin immunoprecipitation by RNA purification in combination with deep sequencing or chromatin immunoprecipitation by RNA purification in combination with mass spectrometry, identification of direct RNA-interacting proteins (iDRiP), R-ChIP for robust mapping of R-loops genome-wide, and DNA-RNA immunoprecipitation combined with deep sequencing,27,33,64-66 enable researchers to map global binding of a particular lncRNA in the genome and identify the associated protein complexes. These studies aid in the investigation of the molecular mechanisms by which lncRNAs access and regulate the genome in high resolution. Furthermore, refined CRISPR-Cas9–mediated genome editing of lncRNA and targeting of RNA-mediated structures (eg, R-loops or triplex) at specific genomic locations will provide important functional links and validations. Eventually, it is anticipated that increasing mechanistic understanding of lncRNA-driven topological genome organization will solidify its link with HSPC regulation and leukemogenesis in the next few years.
Acknowledgments
This work was supported by grants from the National Institutes of Health (National Institute of Diabetes and Digesitive and Kidney Diseases grant R01DK110108, National Cancer Institute grant R01CA204044, and National Heart, Lung and Blood Institute grant R01HL141950 [S.H.]; National Cancer Institute grant R01CA172408, National Heart, Lung and Blood Institute grant R01HL145883, and National Heart, Lung and Blood Institute grant R01HL141950 [M.X.]; and National Heart, Lung and Blood Institute grant R01HL144712 [Y.Q.]), the Cancer Prevention and Research Institute of Texas (grant RP200242 [M.X.]), and the Four Diamonds Fund (S.H.).
Authorship
Contribution: Y.Q., M.X., and S.H. wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Suming Huang, Pennsylvania State University College of Medicine, 500 University Dr, H085, Room T3402, Hershey, PA 17033; e-mail: shuang4@pennstatehealth.psu.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal