Abstract
Mutations in genes encoding RNA splicing factors were discovered nearly 10 years ago and are now understood to be among the most recurrent genetic abnormalities in patients with all forms of myeloid neoplasms and several types of lymphoproliferative disorders, as well as subjects with clonal hematopoiesis. These discoveries implicate aberrant RNA splicing, the process by which precursor RNA is converted into mature messenger RNA, in the development of clonal hematopoietic conditions. Both the protein and the RNA components of the splicing machinery are affected by mutations at highly specific residues, and a number of these mutations alter splicing in a manner distinct from loss of function. Importantly, cells bearing these mutations have now been shown to generate mRNA species with novel aberrant sequences, some of which may be critical to disease pathogenesis and/or novel targets for therapy. These findings have opened new avenues of research to understand biological pathways disrupted by altered splicing. In parallel, multiple studies have revealed that cells bearing change-of-function mutation in splicing factors are preferentially sensitized to any further genetic or chemical perturbations of the splicing machinery. These discoveries are now being pursued in several early-phase clinical trials using molecules with diverse mechanisms of action. Here, we review the molecular effects of splicing factor mutations on splicing, the mechanisms by which these mutations drive clonal transformation of hematopoietic cells, and the development of new therapeutics targeting these genetic subsets of hematopoietic malignancies.
Introduction
Genomic, functional, and clinical studies over the last 10 years have identified that the RNA splicing machinery is frequently targeted by mutations in clonal hematopoietic conditions, including all forms of myeloid neoplasms,1-3 chronic lymphocytic leukemia (CLL),4,5 clonal hematopoiesis (CH),6-9 and mantle cell lymphoma (MCL).10 RNA splicing is the enzymatic process whereby segments of nucleotides from precursor messenger RNA (pre-mRNA) are removed while the remaining ends of RNA are ligated to generate mature RNA sequences. The vast majority of genes in the human genome undergo alternative splicing to generate multiple potential protein-encoding mature mRNAs from a single gene. As such, RNA splicing is well established to mediate proteome diversity. In addition, by generating mRNA species that are targeted for degradation and regulating the expression of isoforms of non–protein-coding RNAs, RNA splicing is also an essential regulator of gene expression.
Given that the splicing machinery regulates the expression and sequences of RNAs encoded from so many genes functioning in a multitude of cellular processes, understanding the effects of these mutations has represented an exciting challenge. In addition, the discovery that many of the mutations affecting RNA splicing factors confer a change of function, distinct from loss of function,11-13 provides a promising possibility for developing novel therapeutic approaches for patients affected by hematologic malignancies with these mutations. Here, we review current knowledge of mutations in RNA splicing factors, their molecular effects on splicing and other cellular processes, and potential therapeutic opportunities created by these mutations.
RNA splicing regulation
RNA splicing is a nuclear process catalyzed by the spliceosome, a metalloribozyme consisting of 5 small nuclear ribonucleoproteins (snRNPs; U1, U2, U4, U5, and U6 snRNPs), each of which contains its own small nuclear RNA (snRNA) complexed to a group of proteins, and >200 related proteins (reviewed recently by Wahl and Lührmann14,15 ). Sequences embedded within pre-mRNA base pair with snRNAs to recruit the spliceosome to the 5' splice site (5'ss; located at the 5' end of a sequence to be removed), the 3'ss (located at the 3' end of a sequence to be removed), the branch-point sequence (BPS; located upstream of the 3'ss), and the polypyrimidine tract (located between the BPS and the 3'ss) (Figure 1A-B). Many of the genetic alterations influencing splicing affect proteins involved in the initial steps of spliceosome assembly (Figure 1C). Spliceosome assembly is achieved by the U1 snRNP binding to the 5'ss, splicing factor 1 (SF1) binding the BPS, and the U2 auxiliary factor (U2AF) complex binding the polypyrimidine tract and 3'ss. The likelihood that the spliceosome recognizes a splice site is influenced by the binding of auxiliary splicing factors that strengthen or repel the recruitment of the spliceosome.16 These auxiliary factors include members of the serine/arginine (SR) protein family, which generally promote splicing by recognizing specific sequences in pre-mRNA named exonic and intronic splicing enhancers (Figure 1D). In contrast, heterozygous nuclear ribonucleoproteins (hnRNPs) generally suppress splicing by interacting with exonic and intronic splicing silencers.17 Additionally, most of splicing occurs cotranscriptionally, and factors that influence the rate of RNA polymerase II elongation may modify splicing outcomes by influencing the likelihood of splice sites being recognized.
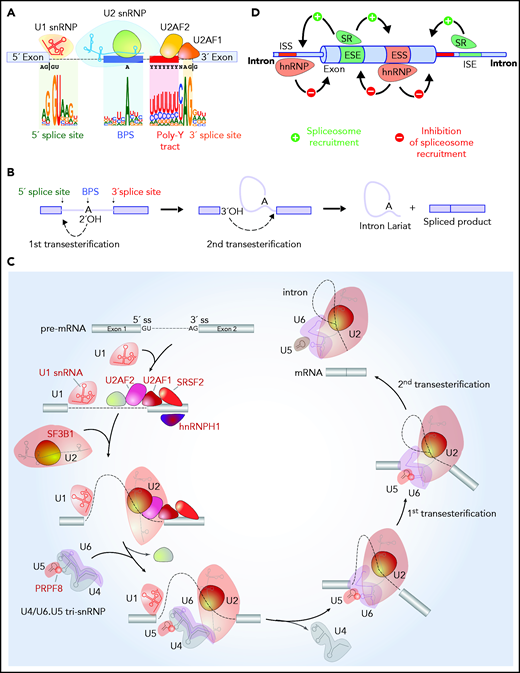
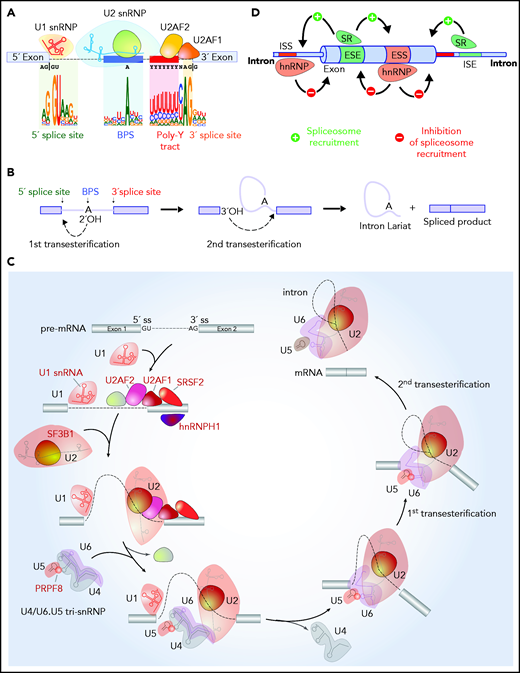
RNA splicing catalysis, splicing regulation, and location of splicing factors mutated in hematologic malignancies in the splicing process. (A) Sequences embedded within premature RNA serve to recruit the spliceosome and include the 5' and 3' splice sites (which are most commonly GU and AG dinucleotides, respectively), the BPS, and polypyrimidine (poly Y) tract. The branch-point nucleotide is most commonly an adenosine nucleotide as shown, but other nucleotides can occasionally serve as branch points, and it is not uncommon for introns to have multiple branch points. (B) Although splicing requires several hundred proteins, the core steps of splicing catalysis consist of 2 sequential transesterification reactions as shown. RNA splicing is initiated when the branch nucleotide performs a nucleophilic attack of the 5'ss, resulting in the formation of an intron lariat intermediate with a 2', 5'-phosphodiester linkage. This is followed by a 5'ss-mediated attack on the 3'ss, leading to the removal of the intron lariat and the formation of the spliced RNA product. (C) The enzymatic steps of splicing are carried out by groups of proteins complexed with snRNAs termed snRNPs. Factors labeled in red in this diagram under recurrent mutations in patients with hematologic malignancies. Splicing is initiated with binding of the U1 snRNP binds the 5'ss, SF1 to the BPS, (iii) U2AF2 to the polypyrimidine tract, and (iv) U2AF1 to the 3ss. These interactions enhance recruitment of U2 snRNP to the BPS. SF3B1, a component of U2 snRNP, is involved in the binding to the BPS. The preassembled U4/U6.U5 tri-snRNP complex joins and the U1/U4 snRNPs are released to form a catalytically active complex of the spliceosome, which catalyze the first and second esterification reactions, respectively, and mediate excision of the intron and ligation of the proximal and distal exon to synthesize mature mRNA. (D) Beyond splice sites, BPS, and the poly Y tract, additional sequences located within introns and exons serve to recruit auxiliary splicing factors, that interact with the spliceosome and promote or repress spliceosome function. These are termed ESEs or exonic splicing silencers (ESSs), respectively) or intronic splicing enhancers or silencers (ISEs or ISSs, respectively). Splicing regulatory proteins known as SR or hnRNP proteins most commonly enhance or repress spliceosome recruitment, respectively, as illustrated.
RNA splicing catalysis, splicing regulation, and location of splicing factors mutated in hematologic malignancies in the splicing process. (A) Sequences embedded within premature RNA serve to recruit the spliceosome and include the 5' and 3' splice sites (which are most commonly GU and AG dinucleotides, respectively), the BPS, and polypyrimidine (poly Y) tract. The branch-point nucleotide is most commonly an adenosine nucleotide as shown, but other nucleotides can occasionally serve as branch points, and it is not uncommon for introns to have multiple branch points. (B) Although splicing requires several hundred proteins, the core steps of splicing catalysis consist of 2 sequential transesterification reactions as shown. RNA splicing is initiated when the branch nucleotide performs a nucleophilic attack of the 5'ss, resulting in the formation of an intron lariat intermediate with a 2', 5'-phosphodiester linkage. This is followed by a 5'ss-mediated attack on the 3'ss, leading to the removal of the intron lariat and the formation of the spliced RNA product. (C) The enzymatic steps of splicing are carried out by groups of proteins complexed with snRNAs termed snRNPs. Factors labeled in red in this diagram under recurrent mutations in patients with hematologic malignancies. Splicing is initiated with binding of the U1 snRNP binds the 5'ss, SF1 to the BPS, (iii) U2AF2 to the polypyrimidine tract, and (iv) U2AF1 to the 3ss. These interactions enhance recruitment of U2 snRNP to the BPS. SF3B1, a component of U2 snRNP, is involved in the binding to the BPS. The preassembled U4/U6.U5 tri-snRNP complex joins and the U1/U4 snRNPs are released to form a catalytically active complex of the spliceosome, which catalyze the first and second esterification reactions, respectively, and mediate excision of the intron and ligation of the proximal and distal exon to synthesize mature mRNA. (D) Beyond splice sites, BPS, and the poly Y tract, additional sequences located within introns and exons serve to recruit auxiliary splicing factors, that interact with the spliceosome and promote or repress spliceosome function. These are termed ESEs or exonic splicing silencers (ESSs), respectively) or intronic splicing enhancers or silencers (ISEs or ISSs, respectively). Splicing regulatory proteins known as SR or hnRNP proteins most commonly enhance or repress spliceosome recruitment, respectively, as illustrated.
The core enzymatic steps of splicing consist of 2 sequential transesterification reactions (Figure 1B). The branch-point nucleotide performs a nucleophilic attack resulting in the formation of an intron lariat. This is followed by a 5′ss-mediated attack on the 3′ss, leading to the removal of the intron lariat and the formation of the spliced RNA product. Excellent reviews of the entire steps of the splicing process14,15 and structural features of the spliceosome18,19 have been written recently.
Recurrently mutated splicing factors in hematologic malignancies and effects on splicing
SF3B1 mutations
The most commonly mutated splicing factor across cancers is SF3B1,20,21 an essential splicing factor and member of the U2 snRNP complex. Mutations in SF3B1 define a distinct clinical entity of myelodysplastic syndrome (MDS), MDS with ring sideroblasts (MDS-RS),1,2,22 while also being among the commonly mutated genes in CLL4,5 and uveal melanoma23-25 (Figure 2A). As part of the U2 snRNP complex, the N terminus of SF3B1 interacts with U2AF heterodimer, while its C-terminal HEAT (Huntington, elongation factor 3, PR65/A, TOR) domain interacts with the BPS and surrounding pre-mRNA sequences to promote binding of the U2 snRNA to the branch point.26,27 SF3B1 mutations mostly occur as heterozygous point mutations at restricted residues within its HEAT domain.2,4,20,22-25
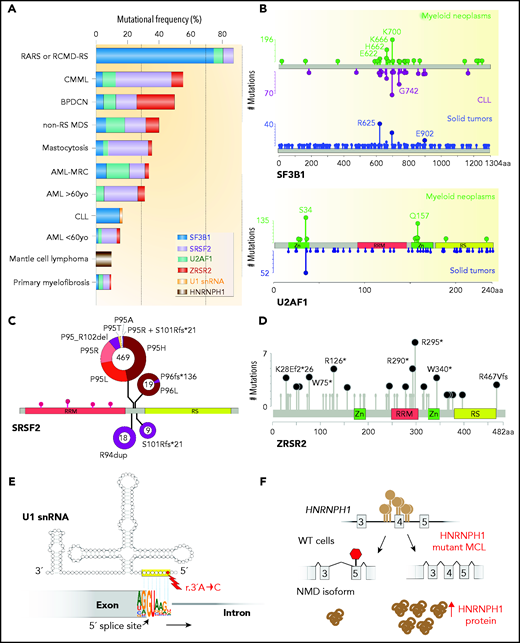
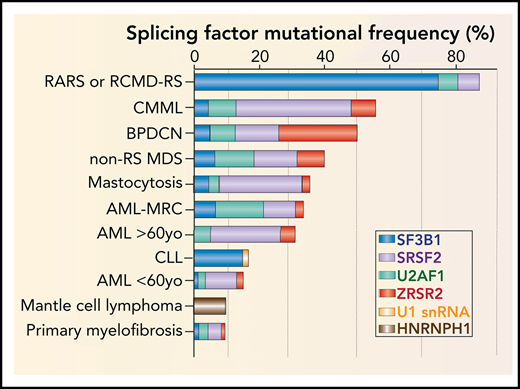
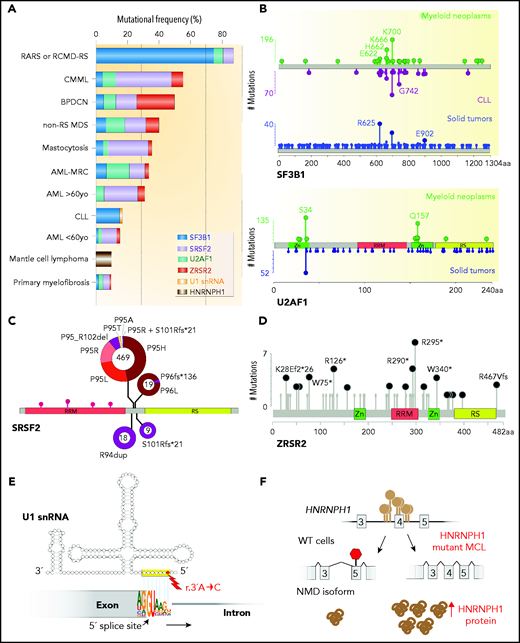
Frequency, genomic characteristics, and effects on splicing of RNA splicing factor mutations seen in hematologic malignancies. (A) Histogram of mutations in the most commonly mutated RNA splicing factors across hematologic malignancies. AML-MRC, acute myeloid leukemia with myelodysplasia-related changes; BPDCN, blastic plasmacytoid dendritic cell neoplasm; CLL: chronic lymphocytic leukemia; CMML, chronic myelomonocytic leukemia; RARS, refractory anemia with ring sideroblasts; RCMD-RS, refractory cytopenia with multilineage dysplasia and ring sideroblasts. (B) Location and relative frequency of mutations in SF3B1 and U2AF1 in myeloid neoplasms, CLL, and solid tumors. (C) Location and relative frequency of mutations in SRSF2 with indication of exact amino acid substitutions at mutated residues. (D) Location and relative frequency of mutations in ZRSR2 displaying known frameshift and insertion/deletion mutations only. (E) Location of recurrent mutations in the gene (or genes) encoding the U1 snRNA affect its third nucleotide, which makes critical base pairs with the 5' splice site. (F) The most frequent mutations in HNRNPH1 cluster in the introns surrounding exon 4 and promote inclusion of this exon. HNRNPH1 mutations in this region occur entirely as single-nucleotide point mutations (as indicated by the brown lollipops). Repression of exon 4 induces a NMD-inducing isoform of HNRNPH1, while inclusion of exon 4 promotes stable HNRNPH1 expression. WT, wild-type.
Frequency, genomic characteristics, and effects on splicing of RNA splicing factor mutations seen in hematologic malignancies. (A) Histogram of mutations in the most commonly mutated RNA splicing factors across hematologic malignancies. AML-MRC, acute myeloid leukemia with myelodysplasia-related changes; BPDCN, blastic plasmacytoid dendritic cell neoplasm; CLL: chronic lymphocytic leukemia; CMML, chronic myelomonocytic leukemia; RARS, refractory anemia with ring sideroblasts; RCMD-RS, refractory cytopenia with multilineage dysplasia and ring sideroblasts. (B) Location and relative frequency of mutations in SF3B1 and U2AF1 in myeloid neoplasms, CLL, and solid tumors. (C) Location and relative frequency of mutations in SRSF2 with indication of exact amino acid substitutions at mutated residues. (D) Location and relative frequency of mutations in ZRSR2 displaying known frameshift and insertion/deletion mutations only. (E) Location of recurrent mutations in the gene (or genes) encoding the U1 snRNA affect its third nucleotide, which makes critical base pairs with the 5' splice site. (F) The most frequent mutations in HNRNPH1 cluster in the introns surrounding exon 4 and promote inclusion of this exon. HNRNPH1 mutations in this region occur entirely as single-nucleotide point mutations (as indicated by the brown lollipops). Repression of exon 4 induces a NMD-inducing isoform of HNRNPH1, while inclusion of exon 4 promotes stable HNRNPH1 expression. WT, wild-type.
A number of studies have performed bioinformatic analyses of patient samples, induced pluripotent stem cells derived from patient samples, leukemia cell lines, and mouse models to identify global splicing change associated with SF3B1 hotspot mutations. Each of these studies has converged on the finding that cells containing SF3B1 mutations have increased expression of mRNAs utilizing aberrant branch-point nucleotides resulting in use of an aberrant (or “cryptic”) 3'ss.13,28 SF3B1 mutant cells tend to use cryptic 3'ss, which are most frequently 10 to 30 bp upstream of the canonical AG dinucleotide. Since the majority of such sites are located at nucleotide distances from the annotated stop codon that are not multiples of 3, a large fraction of the aberrant transcripts generated in SF3B1 mutant cells are predicted to generate premature termination codons (PTCs) subject to nonsense-mediated decay (NMD). RNA-sequencing analysis on SF3B1 mutant MDS-RS patient samples identified alternative 3'ss usage in the iron transporter ABCB7 transcript and consequent generation of a PTC.28 Of note, ABCB7 is specifically downregulated in refractory anemia with ring sideroblasts compared with other MDS subtypes.29,30 NMD-induced downregulation of ABCB7 may explain the strong association of SF3B1 mutations in MDS refractory anemia with ring sideroblasts. In addition, SF3B1 mutants promote NMD of transcripts encoding MAP3K731 and the protein phosphatase 2A subunit PPP2R5A32 due to cryptic 3'ss usage. These mis-splicing events have been functionally characterized in their contribution to malignancies.
Despite the recurrent nature of cryptic 3'ss selection, it is important to note that a number of splicing changes generated by mutant SF3B1 are not aberrant 3'ss events. For example, mutations in SF3B1 lead to aberrant inclusion of a PTC-containing exon (a so-called poison exon) in BRD9 due to aberrant branch-point usage.33 Similarly, SF3B1 mutant cells harbor a number of frame-preserving splicing changes that have been largely uncharacterized.33 Thus, one major area of research related to mutations in SF3B1 and other RNA splicing factors is to systematically catalog the splicing changes within these cells and determine the functional importance of these altered mRNAs to the development of distinct cancer types where SF3B1 mutations are seen. One additional area of research is the application of long-read RNA sequencing technologies to provide greater resolution of full-length RNA transcripts in cells with and without SF3B1 and other splicing factor mutations. This has initially been performed using Oxford nanopore sequencing of CLL patients with SF3B1 mutations using a novel methodology for full-length differential isoform detection.34
In addition to attempts to identify splicing changes created by mutant SF3B1, there have also been efforts to understand the molecular basis by which mutations in SF3B1 alter branch-point usage and/or 3'ss selection. To this end, a set of recent studies by the Manley group suggest that mutations in SF3B1 result in aberrant splicing by disrupting physical interaction of SF3B1 with a spliceosomal protein known as SUGP1.35,36 While the authors were able to show that depletion of SUGP1 mimics splicing changes seen with mutations in SF3B1, it is not clear how or why SUGP1 loss would alter splicing in this way. It is hypothesized that SUGP1 may interact with RNA helicases responsible for allowing the U2 snRNP complex to identify the correct branch-point site. However, further studies are needed to evaluate this hypothesis. The more recent discovery of cancer-associated SUGP1 mutations may further motivate this effort.36
Although most SF3B1 mutations occur within its HEAT domains, intriguingly, distinct mutated residues in SF3B1 appear to be associated with specific cancer types and even specific subtypes of myeloid neoplasms (Figure 2B). For example, R625 substitutions in SF3B1 appear to be strikingly restricted to melanomas and nearly absent in hematologic malignancies.20,23-25 Moreover, while K700E substitution is the most common SF3B1 mutation in MDS-RS, substitutions at the K666 residue occur in ∼1.5% MDS-RS and are distinctly associated with high-risk MDS and acute myeloid leukemia (AML).37 Currently, the basis for these allele-specific effects of distinct SF3B1 mutations are unknown. It is hypothesized that each hotspot mutation may induce unique structural effects on SF3B1, thereby resulting in allele-specific differences in how the U2 snRNP contacts RNA and/or interacts with associated proteins. It also important to note that each SF3B1 mutation may have its own unique set of mutational co-occurrences or exclusivities that may also relate to allele-specific effects on splicing or clinical phenotypes.
U2AF1 and U2AF2 mutations
Along with the SF3b complex, the U2AF heterodimeric complex is critical in delineating the 3'ss and also affected by mutations in hematologic malignancies. The larger subunit of the complex, U2AF2 (also known as U2AF65), binds the polypyrimidine tract between the branch point and the 3'ss, while the smaller subunit, U2AF1 (also known as U2AF35), recognizes the consensus AG dinucleotide at the 3'ss and intron-exon boundary38-40 (Figure 1A). While both U2AF1 and U2AF2 are mutated in myeloid neoplasms, mutations in U2AF1 are far more common and mostly occur at the S34 or Q157 residues located within the first and second zinc-finger domains of U2AF1, respectively1,3 (Figure 2A-B). Interestingly, U2AF1 mutants result in allele-specific alterations in cassette exon usage in a manner dependent on the sequences surrounding the AG dinucleotide at the 3'ss.12 U2AF1S34F/Y mutant cells exhibit preferential use of cassette exons where the 3'ss bears a C or A at the −3 position to the AG while repressing exons bearing a T at the −3 position. In contrast, U2AF1Q157P/R mutations induce recognition of 3'ss bearing a G immediately following the AG dinucleotide while repressing exons bearing an A at the +1 site.12
Within hematologic malignancies, U2AF1 and U2AF2 mutations are largely restricted to myeloid neoplasms and are more often associated with high-risk MDS and AML.3,41,42 U2AF1 mutants alter differential splicing of many genes that affect various biological pathways, including DNA damage response (ATR and FANCA), epigenetic regulation (H2AFY, ASXL1, BCOR, and DNMT3B), apoptosis (CASP8), and innate immune signaling (IRAK4).12,43,44 It has been noted that some of the genes whose transcripts are misspliced by mutant U2AF1 are also recurrently mutated in MDS/AML,43 suggesting that aberrant splicing of these genes may be relevant to myeloid malignancies. One intriguing U2AF1 mutant–induced event is a mutually exclusive exon in H2AFY encoding macroH2A1,12,43 a H2A histone variant. MacroH2A1 has 2 isoforms macroH2A1.1 and macroH2A1.2, due to alternative splicing of mutually exclusive exons. U2AF1S34F-induced aberrant splicing decreases the expression of the isoform encoding macroH2A1.1. Interestingly, knockdown of macroH2A1.1 in hematopoietic progenitor cells resulted in impaired erythroid and granulomonocytic differentiation that phenocopied the differentiation blocks observed in U2AF1 mutant MDS cases.45 As with mutations in SF3B1, it is currently unclear which splicing events created by mutated U2AF1 or U2AF2 may promote leukemia development. One such event observed in U2AF1S34F-associated MDS/AML is mutant induced expression of a long isoform of IRAK4 (IRAK-L) that retains exon 4 containing the N-terminal death domain. This active isoform that is required for leukemic growth interacts with MyD88 resulting in activation of NF-κB and MAPK. Clinically, expression of IRAK-L is associated with poor prognosis in AML.44
Although far less common than mutations in U2AF1, U2AF2 is affected by recurrent hotspot mutations, most of which cluster within the first of its 2 RNA recognition motif domains. These mutations (such as U2AF2 G176 and L187 mutations) are predicted to alter binding of U2AF2 to the polypyrimidine tract,46 but few studies have charted the effects of U2AF2 mutations on splicing alterations within cells to date.
SRSF2 mutations
Beyond mutations in core RNA splicing factors, a number of auxiliary splicing factors are mutated in hematologic malignancies, the earliest of which to be identified were mutations in SRSF2.1 SRSF2 mutations are present in 10% to 14% of patients with AML, 20% to 30% of patients with MDS, and ∼50% of chronic myelomonocytic leukemia patients1,42,47 (Figure 2A). Similar to mutations in U2AF1, mutations in SRSF2 are associated with higher-risk forms of MDS and AML.3,41,42 Moreover, although mutations in U2AF1 and SRSF2 are each identified only in 3% of healthy individuals with CH aged >70 years,48 their detection rises to 10% of younger individuals whose CH converts to overt AML,8,9 suggesting that mutations in SRSF2 or U2AF1 are associated with a high risk of AML transformation.
SRSF2 is a member of the serine/arginine-rich (SR) protein family that contributes to both constitutive and alternative splicing to promote exon recognition by binding to exonic splicing enhancer (ESE) sequences within pre-mRNA through its RNA recognition motif domain.49-52 Work from our group and others has identified that while SRSF2 efficiently recognizes CCNG and GGNG sequences in mRNA,53 leukemia-associated SRSF2 mutations alter SRSF2 binding to RNA in a sequence-specific manner by reducing its ability to recognize G-rich ESE sequences while increasing its avidity to C-rich sequences.11,54 This leads to enhanced inclusion of exons with CCNG sequence and skipping of those with GGNG sequence. Of note, P95 mutations do not affect protein-protein interactions as far as is understood to date.54
The transcriptional and biological effects of specific amino acid substitution at SRSF2 P95 need to be investigated further, as genomic data within the context of CH suggest that P95R substitutions provide hematopoietic stem cells with a larger selective advantage compared with P95H or P95L substitutions55 (Figure 2C). In addition, rare non-hotspot SRSF2 mutations are recurrently detected in hematologic malignancies, and like their hotspot counterparts, they cause a diversity of splicing changes, with cassette exons representing the most common differentially spliced event. Most non-hotspot SRSF2 mutations also alter ESE preference; however, each mutation induces a unique missplicing program suggestive of different functional consequences.56
Many genes differentially spliced by mutant SRSF2 have known importance in myeloid malignancies. For example, SRSF2 mutations promote the inclusion of a highly conserved poison exon within EZH2 that triggers its NMD and consecutive impairment of hematopoietic differentiation.11 Of note, EZH2 loss-of-function mutations are common in MDS and mutually exclusive with SRSF2 mutations.42 Also recurrently misspliced in SRSF2 mutant cells is the gene encoding caspase-8,31 a cysteine protease that in addition to its role in apoptosis is a key activator of NF-κB signaling.57-61 SRSF2P95H mutation represses a cassette exon of caspase-8, resulting in an mRNA encoding a truncated caspase-8 protein lacking the C-terminal catalytic domains.31 This specific isoform detected in SRSF2P95H-mutant cells is distinct from all those previously described62,63 and does not affect cell death but strongly induces NF-κB signaling.31 Interestingly, observation of the shared effects of mutations in SRSF2, SF3B1, and U2AF1 on increased NF-κB signaling has led to a suggestion that activation of NF-κB may be a convergent effect of splicing factor mutations, potentially explaining the enrichment of each of these mutations in myeloid neoplasms.31,44 Further efforts to dissect the contribution of NF-κB signaling to splicing factor mutant myeloid neoplasms may therefore be important to understand disease pathogenesis and illuminate novel therapeutic approaches for this genetic subset of patients.
It is also important to note that functionally important splicing changes beyond cassette exon splicing occur downstream of mutant SRSF2.11,64,65 For example, SRSF2 mutant cells exhibit intron retention resulting in NMD of mRNA encoding INTS3 (integrator subunit 3), an event that contributes to their malignant transformation. The mechanistic basis for recurrent intron retention events in SRSF2 mutant cells has not been fully explored to date despite their potential pathologic significance.65
Although SRSF2, SF3B1, and U2AF1 mutations are frequently found in leukemias, there is no functional evidence that these mutations can generate overt AML or CLL in vivo independently of other concomitant genetic aberrations. To date, 3 distinct Srsf2P95H conditional knockin mouse models have been generated, each from the endogenous Srsf2 locus, and they have been analyzed using inducible deletion using the transgenic Cre expression with Mx1-cre,11 Vav-cre,65,66 and Scl1-CreERT.67 With the exception of one model,67 on its own, hematopoietic expression of Srsf2P95H resulted in a nonproliferative MDS-like phenotype. Conditional expression of U2af1S34F, both from the endogenous mouse U2af1 locus68 and from a transgenic locus,69 also generates a nonproliferative MDS-like phenotype. Finally, expression of the Sf3b1K700E mutation alone throughout the hematopoietic system (using Mx1-cre) has been shown to confer an MDS-like disorder with impaired erythroid differentiation but no competitive or proliferative advantage.31,70 Moreover, expression of Sf3b1K700E within B cells specifically (using CD19-cre) impairs B-cell lymphopoiesis on its own.32,71 However, there is accumulating evidence that these mutations can promote disease development in specific genetic contexts. For example, there is a frequent co-occurrence of mutations in IDH2 and SRSF2 in AML and coexpression of mutant Idh2 and Srsf2 in mice induces aberrant splicing together with epigenomic alterations and lethal myelodysplasia with proliferative features in vivo.65 Similarly, while introduction of the Sf3b1K700E mutation within B cells in mice does not alter B-cell proliferation or numbers, it promotes lymphomagenesis when coexpressed with MYC in B cells.72 Deletion of Atm has also been shown to collaborate with B-cell–restricted expression of Sf3b1K700E to generate CLL in vivo.71 Future work to determine if splicing factor mutations are required for the survival of established malignancies will be important in understanding the potential utility of targeting mutant splicing factors.
ZRSR2 mutations
Within most species, RNA splicing is catalyzed by 2 parallel machineries, the major and the minor spliceosome.73,74 The majority of introns (U2-type introns) are recognized by the major spliceosome, whereas <0.5% of genes contain introns (U12-type introns) marked by highly conserved sequences at their 5' and 3' ends that are excised by a separate spliceosome known as the “minor spliceosome.” Of the recurrently mutated splicing factors in patients with hematologic malignancies, ZRSR2 is the only factor subjected to recurrent mutations thought to primarily function in the minor spliceosome.75 In contrast to mutations in SF3B1, SRSF2, or U2AF1, which are affected by heterozygous hotspot mutations, the X-chromosome–encoded ZRSR2 is enriched for nonsense and frameshift mutations throughout its open reading frame, consistent with loss of function (Figure 2D). As such, ZRSR2-mutant MDS has a male predominance.1 Intriguingly, ZRSR2 mutations are also quite frequent in the subtype of AML known as blastic plasmacytoid dendritic cell neoplasms,76 an aggressive myeloid neoplasm with known male predominance (Figure 2A).
Although ZRSR2’s precise role in splicing is not completely understood, ZRSR2 is an RNA-binding protein (RBP) that is thought to primarily interact with the 3'ss of U12-type introns. RNA-sequencing analysis of ZRSR2-mutant or deleted cells by Madan et al demonstrated that ZRSR2 mutations primarily altered splicing of U12-type introns by inducing minor intron retention.75 While a number of minor intron-containing genes have well known importance in cancer,77 the functional importance of disrupting their expression by minor intron retention is not immediately clear. Thus, the biological basis for enrichment of ZRSR2 mutations in myeloid neoplasms remains to be clarified.
snRNA mutations
In addition to frequent mutations in protein components of the spliceosome, recent work by the International Cancer Genome Consortium has identified recurrent hotspot mutations in the RNA components of the spliceosome known as snRNAs. To date, snRNA mutations have been identified in genes encoding the U1 and U11 snRNAs,78,79 which are snRNAs responsible for recognition of the 5'ss in the major and minor spliceosomes, respectively.
The discovery of mutations in snRNA genes was quite unexpected, as most snRNAs are encoded by many nearly identical genes and arrays of gene copies throughout the genome in addition to the presence of numerous nearly identical snRNA pseudogenes. Thus, snRNA mutations occur within repetitive elements of the genome, making identifications of bona fide somatic mutations in genes encoding snRNAs challenging. Nonetheless, Shuai et al identified recurrent U1 and U11 snRNA mutations in CLL, diffuse large B-cell lymphoma, and sonic hedgehog–type medulloblastoma in addition to other cancer types. These mutations cluster at the third base within the U1 and U11 snRNAs, which base pairs to the 5' splice site (Figure 2E). Consistent with this, RNA-sequencing of isogenic cells and CLL samples bearing U1 snRNA mutations has identified changes in 5'ss usage.78,79 Future efforts to identify transcriptionally active snRNA genes in cancers will be critical to help illuminate other potential snRNA alterations.
HNRNPH1 mutations
Until recently, recurrent mutations in RNA splicing factors were not known in B-cell malignancies beyond CLL. However, recent whole-genome sequencing of MCL by Pararajalingam et al identified recurrent mutations in the RBPs DAZAP1, EWSR1, and HNRNPH1 in 5%, 3%, and 10% of MCL patients, respectively.10 Mutations in HNRNPH1 are particularly interesting, as they occur in and around a single exon within HNRNPH1 and serve to promote an isoform of HNRNPH1 that escapes NMD (Figure 2F). As such, MCL-associated HNRNPH1 mutations increase HNRNPH1 mRNA and protein expression, suggesting a proto-oncogenic role for HNRNPH1. Interestingly, these data are consistent with recent CRISPR screens targeting poison exons in cancer, which also uncovered a strong advantage for skipping HNRNPH1’s poison exon.80 HNRNPH1 is a splicing factor and member of the hnRNP family of RBPs, which are generally thought to repress splicing and oppose the activity of SR proteins.17 Clinically, HNRNPH1 mutations are independently associated with adverse outcome in MCL,10 making future efforts to understand the biological role of HNRNPH1 in MCL critical and highlighting a need to explore means to therapeutically target HNRNPH1.
Mutations in additional spliceosome components
In addition to commonly mutated spliceosomal genes mentioned above, a number of other splicing factors and proteins with roles in RNA metabolism have been identified at lower frequencies in patients with myeloid neoplasms. Chief among these are PRPF8 and LUC7L2. PRPF8 is an evolutionarily conserved core spliceosomal protein essential for pre-mRNA splicing (Figure 1C). PRPF8 is a component of the U5 snRNP and the U4/U6.U5 tri-snRNP. As such, PRPF8 has a central role in RNA splicing catalysis, as it is involved in cross-linking the spliceosome to the 5'ss, the branch point, and the 3'ss (recently reviewed by Grainger and Beggs81 ). PRPF8 is encoded on chromosome 17p13.3 and affected by presumed loss-of-function mutations and occasional copy-number loss due to deletion of chromosome 17.82 Somatic mutations in PRPF8 have been identified in ∼3% of patients with myeloid neoplasms. Loss of PRPF8 results in widespread alteration in cassette exon usage, but the biological and mechanistic role of PRPF8 loss in myeloid neoplasms is not yet clear.
LUC7L2, located on chromosome 7q, encodes a splicing factor protein that is also affected by presumed loss-of-function mutations.83 Unlike PRPF8, LUC7L2’s role in RNA splicing is far less established. LUC7L2 has been reported to interact with some splicing regulators and bind U1 and U2 snRNAs, as shown by crosslinking and immunoprecipitation–sequencing experiments.84 Surprisingly, knockdown of LUC7L2 increased the splicing efficiency of selected introns, suggesting a repressive role of LUC7L2 in selective splice site usage. Reduced expression level of LUC7L2 has been reported in ∼14% MDS patients as a result of LUC7L2 mutations and common del(7q).83 More detailed functional and mechanistic investigations of LUC7L2 are therefore warranted.
While each of the splicing factors noted above are affected by somatic mutations, germline and somatic mutations in the RNA helicase known as DDX41 have been recently reported in patients with MDS and AML.85 Germline hotspot and presumed loss-of-function mutations in DDX41 were discovered in adult familial AML syndrome, characterized by long latency, high-risk MDS/AML, and poor prognosis. DDX41 encodes a DEAD-box type adenosine triphosphate–dependent RNA helicase located on chromosome 5q35; thus, haploinsufficiency of DDX41 is also found in some patients with del(5q). To date, multiple roles for DDX41 have been suggested including a potential role in RNA splicing,85 snoRNA processing,86 as well as ribosomal RNA biogenesis.86 This latter effect of DDX41 mutations is intriguing given the well-established examples of familial MDS/AML predisposition syndromes affecting ribosome biogenesis. Currently, it is unclear which of the proposed roles for DDX41 are critical in myeloid malignancy pathogenesis, and the precise role of DDX41 in RNA splicing is also unclear.
Effects of mutant splicing factors on processes beyond splicing
Emerging data suggest that mutant RNA splicing factors alter biological process in cells beyond RNA splicing. One prominent example is the effect of mutations in splicing factors on the formation of R-loops, the RNA-DNA hybrids that form during transcription when nascent RNA anneals to its complementary DNA template. Interestingly, MDS-associated SRSF2 and U2AF1 mutations have been shown to enhance R-loop formation as a consequence of impairing transcription pause release.87,88 The resulting R-loop elevation triggers the DNA-damage response and subsequent replication stress and Chk1-ATR activation when cells enter S phase (Figure 3A). Importantly, suppression of R-loops by RNASEH1 overexpression partially rescues the compromised phenotype observed in Srsf2P95H-mutant hematopoietic progenitor cells.87 These findings suggest that splicing factor mutations may drive blood disorders in part through excessive R-loop formation.
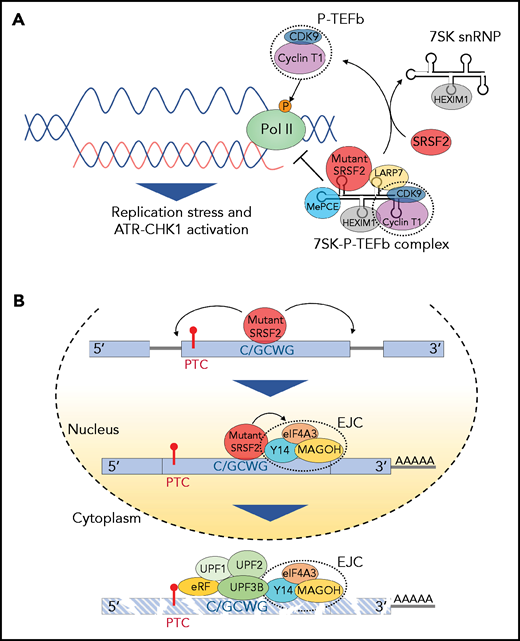
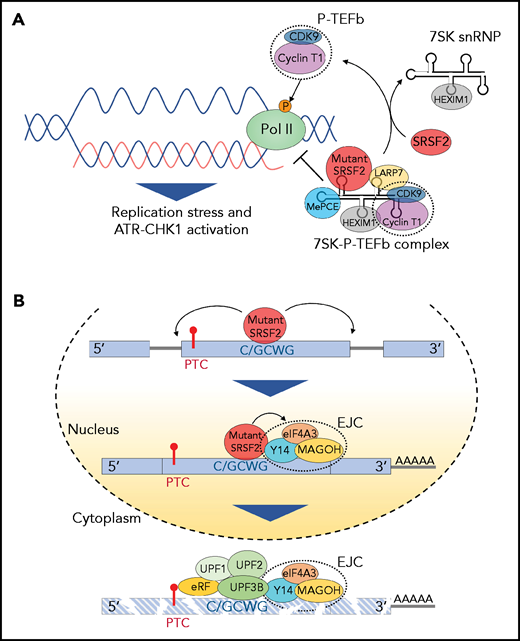
Effect of splicing factor mutations on biological processes beyond RNA splicing. (A) The positive transcription elongation factor complex, P-TEFb, composed of cyclin-dependent kinase 9 (CDK9) and cyclin T1, stimulates synthesis of RNA through phosphorylation of RNA polymerase II (Pol II). However, when bound to 7SK snRNA, HEXIM,1 LARP7, and MePCE (7SK snRNA methyl phosphate capping enzyme), P-TEFb is catalytically inactive and represses transcription by suppressing the release of paused polymerase II. The transition of P-TEFb from repressive to active complexes depends on multiple factors including SRSF2. Mutant SRSF2, however, loses its ability to extract P-TEFb from the 7SK complex due to increased RNA binding. This results in elevated R-loops (nascent RNA-DNA hybrids formed during transcription leaving the nontemplate DNA strand looping out) and subsequent replication stress and activation of the ataxia telangiectasia and Rad3-related protein (ATR)-Chk1 pathway. (B) Mutations at proline 95 residue in SRSF2 change its RNA-binding affinity from G-rich (GGWG) to C-rich (C/GCWG) motifs (W = A/U) inducing transcriptome-wide missplicing events. Several mRNA isoforms promoted by SRSF2 mutants harbor a PTC and are therefore potential targets of NMD. Moreover, SRSF2 mutants further enhance NMD by promoting recruitment of EJC factors (eIF4A3, MAGOH, and Y14) to mRNAs downstream of PTCs within the nucleus. This subsequently enhances the association of several NMD factors (UPF3B, UPF2, and UPF1) to mRNA within the cytoplasm, thereby enhancing mRNA decay.
Effect of splicing factor mutations on biological processes beyond RNA splicing. (A) The positive transcription elongation factor complex, P-TEFb, composed of cyclin-dependent kinase 9 (CDK9) and cyclin T1, stimulates synthesis of RNA through phosphorylation of RNA polymerase II (Pol II). However, when bound to 7SK snRNA, HEXIM,1 LARP7, and MePCE (7SK snRNA methyl phosphate capping enzyme), P-TEFb is catalytically inactive and represses transcription by suppressing the release of paused polymerase II. The transition of P-TEFb from repressive to active complexes depends on multiple factors including SRSF2. Mutant SRSF2, however, loses its ability to extract P-TEFb from the 7SK complex due to increased RNA binding. This results in elevated R-loops (nascent RNA-DNA hybrids formed during transcription leaving the nontemplate DNA strand looping out) and subsequent replication stress and activation of the ataxia telangiectasia and Rad3-related protein (ATR)-Chk1 pathway. (B) Mutations at proline 95 residue in SRSF2 change its RNA-binding affinity from G-rich (GGWG) to C-rich (C/GCWG) motifs (W = A/U) inducing transcriptome-wide missplicing events. Several mRNA isoforms promoted by SRSF2 mutants harbor a PTC and are therefore potential targets of NMD. Moreover, SRSF2 mutants further enhance NMD by promoting recruitment of EJC factors (eIF4A3, MAGOH, and Y14) to mRNAs downstream of PTCs within the nucleus. This subsequently enhances the association of several NMD factors (UPF3B, UPF2, and UPF1) to mRNA within the cytoplasm, thereby enhancing mRNA decay.
In addition to R-loop formation, there is significant interest into whether and how mutations in RNA splicing factors might affect NMD. Although mutations in splicing factors generate mRNA isoforms harboring PTCs due to missplicing, there is a potential that mutant splicing factors may alter the activity and/or specificity of NMD beyond simply generating NMD-inducing mRNA substrates. Recent work from Rahman et al identified that SRSF2P95 mutants, but not wild-type SRSF2, enhance NMD by promoting recruitment of the exon junction complex (EJC) to mRNAs downstream of PTCs within the nucleus.89 This represents the earliest steps of NMD and occurs in a manner dependent on sequence-specific RNA-binding activity of SRSF2 described earlier. The elevated association of SRSF2 mutant with its binding motif enhances deposition of EJCs downstream of the PTC. This architecture then favors the association of key NMD factors to elicit mRNA decay89 (Figure 3B). Further work to evaluate the effect of mutations in other RNA splicing factors on NMD will be important in determining whether other alterations impact NMD beyond effects on splicing.
One additional aspect of RNA processing beyond splicing that may be affected by mutant RNA splicing factors is alterations in mRNA polyadenylation and cleavage. For example, although U1 snRNA mutations appear to be associated with changes in 5'ss usage, it is important to note that the U1 snRNA has also been shown to play a prominent role in suppressing premature cleavage and polyadenylation of RNA (a process termed “telescripting”).90 Given recent data identifying a high frequency of aberrant premature polyadenylation in CLL,91 it will be important to understand if snRNA mutations affect this process. Moreover, work by Park et al suggested that U2AF1S34 mutations alter polyadenylation site usage by interfering with normal interactions between the U2AF complex and the cell’s polyadenylation and cleavage machinery.92 Given the evidence that splicing factors interact with polyadenylation machinery, it will be intriguing to explore whether splicing factor mutations affect intronic polyadenylation in hematological malignancies. Finally, the U2AF1/U2AF2 heterodimer has been recently shown to bind mature mRNAs in the cytoplasm and represses mRNA translation.93 This was an unexpected result as U2AF heterodimer has previously only been known to have a role in RNA splicing within the nucleus. Moreover, in these same studies, the U2AF1 S34F mutation induced translational upregulation and consequent increased secretion of interleukin 8, a chemokine that contributes to cancer progression and is associated with relapsed/refractory AML in humans.93
Therapeutic implications of splicing factor mutations
SF3b binding agents
The discovery of highly recurrent change-of-function mutations in splicing factors in leukemias and CH has generated interest to therapeutically target splicing factor mutant cells. While there are still no data that splicing factor mutations are required for the maintenance of established leukemias, there is abundant evidence that cells bearing hotspot mutations in splicing factors are dependent on otherwise wild-type splicing function. A very strong mutual exclusivity of splicing factor mutations was evident from the initial discovery of mutations in splicing factor mutations in patients with myeloid malignancies.1 This finding suggests potential convergent effects and/or synthetic lethal interactions of these mutations that restricts their co-occurrence. Although several overlapping effects of splicing factor mutations in myeloid neoplasms have been posited (including enhanced generation of R-loops87 and activation of innate immune signaling31 ), no single unifying downstream effect of mutations in SF3B1, SRSF2, U2AF1, and ZRSR2 in myeloid neoplasms is evident to date. In contrast, there are clear functional data demonstrating that coexpression of the most common mutations in SF3B1, SRSF2, or U2AF1 is intolerable in cells.31,94 Similarly, deletion of the wild-type allele in splicing factor mutant cancer cells results in prompt cell death across a number of cancer types bearing mutations in SF3B1,95 SRSF2,96 or U2AF1.97 In contrast, expression of a single wild-type allele encoding these factors is well tolerated.
The above data provide genetic evidence for the dependence of splicing factor mutant cells on otherwise wild-type splicing function and have motivated efforts to use drugs that target splicing catalysis to preferentially eradicate splicing factor mutant cancers. The earliest class of molecules to have been studied in this context include a class of natural products and their synthetic analogs that bind to the SF3b complex and prevent its interaction with the branch point98,99 (Figure 4A). These include the compounds E7107, H3B-8800, pladienolide B, herboxiedene, and spliceostatin A, among other related molecules.100,101 Functional genomic studies have elucidated the specificity of these for the U2 snRNP complex as mutations in SF3B1 (SF3B1R1074H) and an additional U2 snRNP complex member known as PHF5A (PHF5AY36C) confer resistance to these molecules27,102 (Figure 4A). Moreover, crystal26 and cryoelectron microscopy27 structures of the SF3b complex bound to pladienolide B and E7107, respectively, have now been published and reveal that these drugs work by interacting with the branch-point adenosine-binding pocket of SF3B1. Thus, these compounds block U2 snRNP’s recognition of RNA and thereby result in dose-dependent increases in intron retention and cassette exon skipping at thousands of splicing events genome-wide.96,100 Importantly, H3B-8800, a compound currently in phase 1 clinical trials in relapsed/refractory MDS and AML, binds to both wild-type and leukemia-associated mutated SF3B1 equally well. Thus, the basis for the preferential effects of these compounds on leukemia cell lines with splicing factor mutations is the dependence of splicing factor mutant cells on wild-type splicing catalysis (rather than these drugs having a proclivity to binding splicing factor proteins based on their mutational status). As such, preclinical data demonstrate utility for SF3b inhibition for SF3B1,70,100 SRSF2,96 and U2AF1103 mutant leukemia cells to date. Although mouse models of mutant SRSF2, U2AF1, and SF3B1 mutations do not fully recapitulate the individual splicing changes seen in human cells, the shared global impact on RNA splicing across human and mouse mutant cells11,31,68 has made such tractable mouse models particularly valuable for preclinical in vitro and in vivo drug testing. However, given that U2 snRNP function is essential for nearly all cells, the therapeutic index of such drugs remains to be determined. Prior phase 1 clinical trials of E7107 in solid-tumor patients resulted in unexpected ocular toxicities, the basis of which is not yet clear, nor is it clear if similar toxicities would be seen with other SF3b-binding agents.104 Finally, it is important to note that the efficacy of this approach depends on sufficient on-target inhibition of splicing in patients, which will be an important biomarker to assess when evaluating results of the ongoing phase 1 trial of H3B-8800.
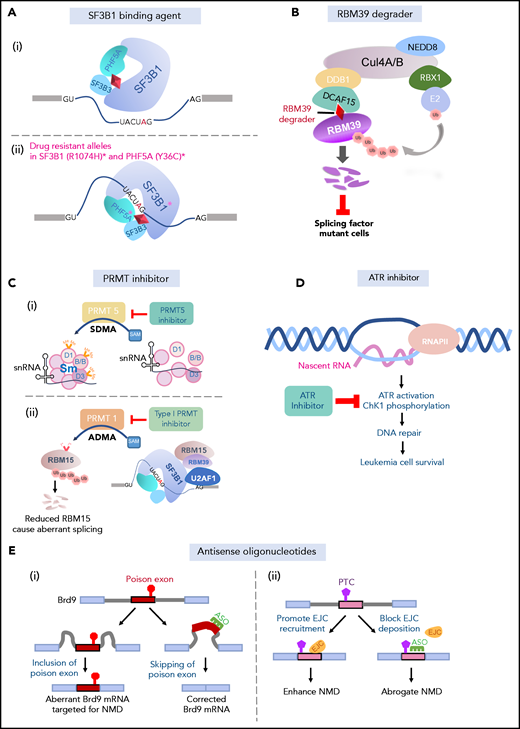
Approaches to targeting RNA splicing. (A) SF3b binding agents physically interact with the branch-point binding pocket of SF3B1, thus blocking its binding with the branch point (i). Specific mutant residues in SF3B1 and PHF5A confer drug resistance to SF3b-binding agents (ii). (B) RBM39 degraders link the E3 ubiquitin ligase complex to RBM39 through the adaptor protein DCAF15, leading to polyubiquitination and subsequent proteosomal degradation of RBM39. Splicing factor–mutant leukemic cells are preferentially sensitive to RBM39 degradation. (C) PRMT5 inhibitors inhibiting PRMT5-mediated symmetric demethylation of arginines (SDMA) on Sm (D1, B/B, D3) proteins, which is required for spliceosome assembly (i). PRMT1 mediates asymmetric demethylation of arginines (ADMA) on RMB15, an RNA-binding protein regulating RNA splicing, among many additional splicing factors. Methylated RBM15 is targeted for polyubiquitination and proteosomal degradation, leading to aberrant splicing (ii). Type 1 PRMT inhibitors may prevent mis-splicing through dampening RBM15 degradation (ii). (D) Elevated R-loop formation in mutant splicing factor cells results in activation of ATR signaling pathway and DNA-damage response. Leukemic cells harboring splicing factor mutations preferentially respond to ATR inhibition. (E) ASOs complementary to the poison exon of Brd9 correct aberrant inclusion of the poison exon (i). ASOs block EJC deposition site on mRNA and prevent recruitment of the EJC downstream of a PTC, thereby preventing NMD induced by splicing factor mutations (ii).
Approaches to targeting RNA splicing. (A) SF3b binding agents physically interact with the branch-point binding pocket of SF3B1, thus blocking its binding with the branch point (i). Specific mutant residues in SF3B1 and PHF5A confer drug resistance to SF3b-binding agents (ii). (B) RBM39 degraders link the E3 ubiquitin ligase complex to RBM39 through the adaptor protein DCAF15, leading to polyubiquitination and subsequent proteosomal degradation of RBM39. Splicing factor–mutant leukemic cells are preferentially sensitive to RBM39 degradation. (C) PRMT5 inhibitors inhibiting PRMT5-mediated symmetric demethylation of arginines (SDMA) on Sm (D1, B/B, D3) proteins, which is required for spliceosome assembly (i). PRMT1 mediates asymmetric demethylation of arginines (ADMA) on RMB15, an RNA-binding protein regulating RNA splicing, among many additional splicing factors. Methylated RBM15 is targeted for polyubiquitination and proteosomal degradation, leading to aberrant splicing (ii). Type 1 PRMT inhibitors may prevent mis-splicing through dampening RBM15 degradation (ii). (D) Elevated R-loop formation in mutant splicing factor cells results in activation of ATR signaling pathway and DNA-damage response. Leukemic cells harboring splicing factor mutations preferentially respond to ATR inhibition. (E) ASOs complementary to the poison exon of Brd9 correct aberrant inclusion of the poison exon (i). ASOs block EJC deposition site on mRNA and prevent recruitment of the EJC downstream of a PTC, thereby preventing NMD induced by splicing factor mutations (ii).
RBM39 degraders
Uncertainties regarding the safety of targeting core spliceosome function underscore the importance of identifying means to perturb splicing through modulation of accessory RNA splicing factors. Fortuitously, recent work by Han et al and Uehara et al identified that aryl sulfonamide molecules (including indisulam, tasisulam, E7820, and chloroquinoxaline sulfonamide) bridge RBM39, an accessory RNA splicing factor, to CRL4-DCAF15 E3 ubiquitin ligase, resulting in proteasomal degradation of RBM39 and dose-dependent splicing defects105,106 (Figure 4B). As with SF3b-binding agents, a series of functional genomic105,107 and structural studies108-110 have revealed the specificity of these molecules for RBM39 and their mechanism of action at subatomic resolution. These more recent studies identify that aryl sulfonamide molecules degrade RBM39 and its paralog, RBM23, the latter of which appears dispensable for cell survival.107 In contrast, RBM39 is required for the survival of a variety of cancer types, and chemical degradation of RBM39 preferentially kills splicing factor mutant leukemias over their wild-type counterparts.111 Interestingly, a series of phase 1 and 2 clinical trials have already been performed with a number of RBM39 degrading compounds in cancer patients before the mechanism of action was known.112-114 Although the safety and maximum tolerated dose of these molecules was thereby defined, it is unknown if RBM39 degradation or subsequent splicing changes are observed in patients at these doses. Thus, further clinical studies of these molecules in the specific genetic context of spliceosomal mutant cancers with appropriate pharmacodynamic readouts will be essential in determining their potential clinical utility in splicing factor–associated leukemias.
PRMT inhibitors
Posttranslational modification (PTM) of splicing factors regulates spliceosome assembly, subcellular localization, and protein-protein interactions required for efficient splicing (reviewed recently by Fong et al115 ). PTMs known to impact splicing function include lysine phosphorylation116,117 (mediated by a number of kinases including SR protein kinases and Cdc2-like kinase) as well as arginine methylation118,119 (mediated by type I and II protein arginine methyltransferases [PRMTs]). As such, inhibitors of enzymes responsible for these processes are being explored as potential therapeutic approaches for splicing factor mutant cells. Unlike SF3b-binding agents and RBM39 degraders, inhibiting enzymes that place PTMs of splicing proteins affect numerous splicing factors at a multitude of amino acid residues118,119 (Figure 4C) in addition to cellular substrates unrelated to splicing (reviewed recently by Yang and Bedford120 ), making dissection of their precise mechanisms of action complicated. Nonetheless, recent data suggest that the most abundant arginine methyl substrates in leukemia cells are splicing proteins.119 Consistent with this, inhibitors of PRMT5 and type I PRMT enzymes, many of which are currently in phase 1 clinical trials for a number of cancers, preferentially kill splicing factor mutant cells over their wild-type counterparts.119 These data highlight the importance of testing such approaches in the context of myeloid neoplasms bearing hotspot mutations in SF3B1, SRSF2, and U2AF1. At least 2 phase 1/2 trials that include arms to test PRMT5 inhibitors in spliceosomal mutant myeloid neoplasms refractory to standard therapy are ongoing, including PRMT5 inhibitors from Glaxo Smith Klein (GSK3326595; www.clinicaltrials.gov #NCT03614728) and Prelude Therapeutics (PRT543; www.clinicaltrials.gov #NCT03886831).
Targeting downstream effects of splicing factor mutations
In parallel with efforts to generate a synthetic lethal interaction between splicing factor mutations and inhibition of splicing, a number of studies have begun to target downstream dependencies generated by splicing factor mutations. One of the most advanced approaches targets the enhanced generation of R-loops in spliceosomal mutant cells. SF3b inhibition as well as mutations in either U2AF1S34 or SRSF2P95 all result in increased R-loops and subsequent activation of ATR kinase, which, in turn, appears to be required to resolve R-loops88,121 (Figure 4D). Interestingly, cells bearing these alterations have enhanced sensitivity to ATR inhibitors, and there is a synergistic effect of ATR and SF3b inhibition in vitro. These data therefore identify a potential novel druggable dependency of spliceosomal mutant cells on R-loop accumulation and ATR response.98 This concept is now being tested in a phase 1B clinical trial of the ATR inhibitor AZD6738 in patients with MDS and chronic myelomonocytic leukemia who have failed first-line therapy (www.clinicaltrials.gov #NCT03770429). Moreover, these efforts underscore the importance of studying the functional effects of splicing factor mutations in detail, as there may be numerous downstream dependencies generated by these mutations that are not yet well understood.
Oligonucleotide approaches
One of the most clinically successful approaches to therapeutically targeting splicing to date has been the use of antisense oligonucleotides (ASOs) and small molecules to correct pathogenic splicing alterations in monogenic disorders. Whether correction of individual splicing changes generated by splicing factor mutations will have therapeutic impact is not yet clear given the hundreds of altered splicing events and numerous coexisting genetic alterations in these cells. Moreover, delivery of therapeutic oligonucleotides to hematopoietic cells presents an additional technical challenge. Nonetheless, efforts of modulating splicing as well as NMD in splicing factor mutant cancer types are greatly needed to test the requirement of individual mRNA perturbations to disease phenotypes. For example, recent work used ASOs to block aberrant inclusion of a poison exon of BRD9 in SF3B1 mutant uveal melanoma cells33 (Figure 4E). In this context, correction of BRD9 splicing restored BRD9 protein levels and had significant antitumor effect in vitro and in vivo. Whether such a result would be seen in SF3B1-mutant hematopoietic malignancies is unclear, and the therapeutic benefit of correcting similar aberrant splicing events in spliceosomal mutant leukemia will be critical to evaluate.
In addition to the use of ASOs to correct aberrant splicing event, they can also serve to block NMD. As noted earlier, SRSF2 mutations stimulate NMD by promoting EJC recruitment downstream of PTCs on aberrantly spliced mRNAs.89 Importantly, targeted blocking of EJC deposition by ASOs abrogated SRSF2 mutant–mediated NMD of specific transcripts. Whether retaining mRNAs harboring PTCs can rescue hematological phenotypes remains to be addressed, and this presents a potential orthogonal ASO technique to target spliceosomal mutant hematopoietic malignancies.
Conclusions and open questions
The highly recurrent nature of mutations in RNA splicing factors provides strong genetic evidence for a role of altered splicing in driving the development and/or maintenance of clonal hematopoietic disorders. Since the initial discovery of these mutations in myeloid neoplasms and CLL nearly 10 years ago, many questions regarding the biological and therapeutic importance of these alterations remain to be answered. For example, it is still not clear if mutations in RNA splicing factors are required for the maintenance of cancer. This will be critical to determine, as therapeutic approaches targeting cells with these mutations or their downstream events are explored. Similarly, high-throughput functional genomic efforts to systematically describe, evaluate, and prioritize altered splicing events generated by these mutations (eg, via CRISPR, RNAi, and complementary DNA screens) are critically needed. In parallel, detailed functional investigations of individual aberrant splicing alterations are still needed to understand disease pathogenesis and nominate downstream splicing events for novel therapies (such as small molecules or oligonucleotides to correct splicing events). As part of these efforts, it will be important to consider allele-specific mutations in RNA splicing factors or distinct amino acid substitutions at the same residues that may yield unique effects on splicing and be associated with their own clinical phenotypes. While several studies cite that a multitude of individual splicing events likely contribute en mass to drive disease phenotypes, there have been few studies to test this hypothesis, and efforts to stringently evaluate correction of individual splicing events are still nascent. Finally, from a therapeutic perspective, further data are needed to understand whether the various chemical means to inhibit RNA splicing catalysis will effectively modulate splicing in vivo with an acceptable therapeutic index.
Acknowledgments
O.A.-W. is supported by the Evans Foundation, the Henry & Marilyn Taub Foundation, and National Institutes of Health (NIH), National Cancer Institute (1 R01 CA201247-01A1, 1 R01 C`A242020-01A1, and 1 R01 CA251138-01), andthe NIH, National Heart, Lung, and Blood Institute (2R01HL128239-06).
Authorship
Contribution: S.C., S.B., and O.A.-W. wrote the manuscript and created the figures.
Conflict-of-interest disclosure: O.A.-W. has served as a consultant for H3B Biomedicine, Foundation Medicine, Merck, Prelude, and Janssen; is on the scientific advisory board of Envisagenics and AIChemy; and has received prior research funding from H3B Biomedicine and LoxoOncology unrelated to the current manuscript.
Correspondence: Omar Abdel-Wahab, 1275 York Ave, New York, NY 10065; e-mail: abdelwao@mskcc.org.