Key Points
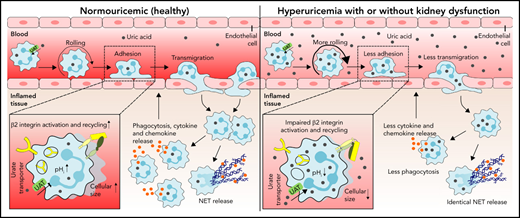
Hyperuricemia suppresses neutrophil adhesion and extravasation during sterile inflammation.
Uric acid impairs neutrophil migration by suppressing β2 integrin activity/recycling and cytoskeletal dynamics upon urate transporter–mediated uric acid uptake.

Abstract
Neutrophils are key players during host defense and sterile inflammation. Neutrophil dysfunction is a characteristic feature of the acquired immunodeficiency during kidney disease. We speculated that the impaired renal clearance of the intrinsic purine metabolite soluble uric acid (sUA) may account for neutrophil dysfunction. Indeed, hyperuricemia (HU, serum UA of 9-12 mg/dL) related or unrelated to kidney dysfunction significantly diminished neutrophil adhesion and extravasation in mice with crystal- and coronavirus-related sterile inflammation using intravital microscopy and an air pouch model. This impaired neutrophil recruitment was partially reversible by depleting UA with rasburicase. We validated these findings in vitro using either neutrophils or serum from patients with kidney dysfunction–related HU with or without UA depletion, which partially normalized the defective migration of neutrophils. Mechanistically, sUA impaired β2 integrin activity and internalization/recycling by regulating intracellular pH and cytoskeletal dynamics, physiological processes that are known to alter the migratory and phagocytic capability of neutrophils. This effect was fully reversible by blocking intracellular uptake of sUA via urate transporters. In contrast, sUA had no effect on neutrophil extracellular trap formation in neutrophils from healthy subjects or patients with kidney dysfunction. Our results identify an unexpected immunoregulatory role of the intrinsic purine metabolite sUA, which contrasts the well-known immunostimulatory effects of crystalline UA. Specifically targeting UA may help to overcome certain forms of immunodeficiency, for example in kidney dysfunction, but may enhance sterile forms of inflammation.
Introduction
Neutrophils play a critical role in acute inflammatory responses such as infection and sterile injury (eg, coronavirus infection, wounds, trauma, or crystal deposition).1,2 During sterile inflammation, neutrophil recruitment from the bloodstream into the inflamed tissue involves several proinflammatory chemokines and cytokines such as CXC motif chemokine ligand 8 (CXCL8), tumor necrosis factor α, and interleukin (IL)-1β, as well as inflammatory changes of the endothelium.3,4 The recruitment of neutrophils is a highly regulated process consisting of several distinct steps, including rolling, adhesion, and crawling and eventually transmigration into the inflamed tissue,3 mechanisms that are mediated by both selectin and integrin interactions with the endothelial surface. Neutrophil slow rolling and adhesion of neutrophils require adhesion molecules of the β2 integrin including lymphocyte function–associated antigen (LFA)-1 (CD11a/CD18, αLβ2) and macrophage-1 antigen (MAC-1, CD11b/CD18, αMβ2).3,5 Mice deficient in the β2 integrin chain CD18 show defects in leukocyte recruitment during inflammatory arthritis6 and peritonitis,7 accompanied by a pronounced increase in leukocyte counts, suggesting an important role for β2 integrins in neutrophil recruitment. This is confirmed in patients with leukocyte adhesion deficiency 1, a disorder with defective or absent CD18. These patients are highly susceptible to developing severe bacterial and fungal infections.8 Upon entry into the inflamed tissue, neutrophil activation exerts various effector functions including phagocytosis, release of proinflammatory mediators and formation of neutrophil extracellular traps (NETs).3,5,9
Uric acid (UA) is a metabolic breakdown product of purine nucleotides. Unlike rodents, humans and higher primates lack the enzyme uricase that degrades UA into the more water-soluble allantoin.10 As a consequence, serum UA levels are 3 times higher in humans. Typical causes of human hyperuricemia (HU), as defined by a serum UA level ≥6.5 mg/dL, include germline mutations in UA transporters as well as various forms of kidney disorders.11 Hyperuricemia is a causal risk factor for gouty arthritis,12 acute UA nephropathy,13 urolithiasis, and kidney stone disease.14 Several in vitro studies also reported proinflammatory effects of UA, including activation of the Akt-PRAS40 pathway and the NLRP3 inflammasome in immune cells,15,16 as well as reactive oxygen species production in nonimmune cells.17 Such proinflammatory effects of UA are consistently documented for its crystalline form.12,18 In contrast, data are less consistent for soluble UA (sUA) that exerts antioxidant properties11 and elicits protective effects in neurodegenerative diseases19 or age-related cancer.
Patients with kidney dysfunction, especially those on dialysis, suffer from infections and inflammation, significantly contributing to overall mortality,20,21 which has been linked to an unresponsiveness of immune cells, including suppressed leukocyte functions and reduced maturation of T helper lymphocytes.22-24 Whether sUA contributes to the immune dysfunction of neutrophils in this context remains unexplored. We hypothesized that HU affects neutrophil recruitment during acute damage- and pathogen-associated molecular patterns (DAMP/PAMP)-induced sterile inflammation (eg, crystals, coronavirus, in kidney dysfunction), whereby sUA impairs β2 integrin–dependent neutrophil recruitment.
Methods and materials
Animal studies
All animal experiments were performed in accordance with the European protection law of animal welfare and upon approval by the local government authorities Regierung von Oberbayern (reference number: ROB-55.2-2532.Vet_02-15-189 and Vet.02-20-224) based on the European Union directive for the Protection of Animals Used for Scientific Purposes (2010/63/EU) and reported according to the Animal Research: Reporting of In Vivo Experiments guidelines.25 Mice were housed in groups of 5 in filter-top cages and had access to food and water. Cages, nest lets, food, and water were sterilized by autoclaving before use. Detailed information on the mouse models including model of HU and/or kidney dysfunction, air pouch model of sterile inflammation, intravital microscopy of the cremaster muscle postcapillary venules, and acute ischemic organ injury can be found in supplemental Methods.
Human study design
The study included 10 patients with chronic kidney disease (CKD)-related HU without dialysis (CKD, CKD stage G2-4), 18 patients with CKD stage G5D that were on hemodialysis with a mean dialysis duration of 57.2 months prior to blood collection (end-stage kidney disease [ESKD]), and 15 healthy individuals without renal impairment (healthy, CKD stage G0) (Table 1). There was no significant difference in age between the groups. We excluded 8 patients due to the intake of immunosuppressive drugs or xanthine oxidase inhibitors. Clinical parameters and the kidney pathologies of the patients are listed in Tables 1 and 2, respectively. The study to obtain whole blood samples from healthy individuals (reference number: 167-12) and patients with kidney disease was approved by the local ethical review board of the medical faculty at the Ludwig-Maximilians-University (LMU) Munich (reference number: 369-15) and carried out in accordance with the declaration of Helsinki for medical research.26 Written informed consent was obtained from all subjects prior to inclusion in the study.
Isolation of human blood neutrophils
Neutrophils were isolated from healthy individuals and patients with kidney dysfunction (CKD/ESKD) before dialysis using standard dextran sedimentation followed by Ficoll-Hypaque density centrifugation procedures.27 Neutrophils were identified by flow cytometry using the antibodies fluorescein isothiocyanate (FITC) anti-human MAC-1 (BioLegend, Fell, Germany), phycoerythrin anti-human CD66b, and allophycocyanin anti-human CD15 (both from eBioscience, Germany). Neutrophils were suspended in RPMI (0.5 × 106 cells per 300 µL or 1 × 106 cells/mL) and seeded onto 8-well microslides (Ibidi, Martinsried, Germany) or 24-well plates in a 5% carbon dioxide atmosphere at 37°C for 30 minutes before stimulation.
Statistical analysis
Statistical analysis was performed using GraphPad Prism 7 software. Data were compared either by Student t test to calculate significance between 2 groups, by 1-way analysis of variance (ANOVA) with Tukey’s post-hoc test between 3 or more groups, or by 2-way ANOVA with Bonferroni’s comparison post-hoc test when using 2 parameters with multiple groups. Data are presented as mean plus or minus standard deviation (SD). Differences were considered significant if P < .05. ns indicates not significant. Sample sizes are indicated in each corresponding figure legend.
Detailed materials and methods are described in supplementary Methods.
Results
Hyperuricemia attenuates neutrophil recruitment during acute crystal- and coronavirus-related sterile inflammation in mice
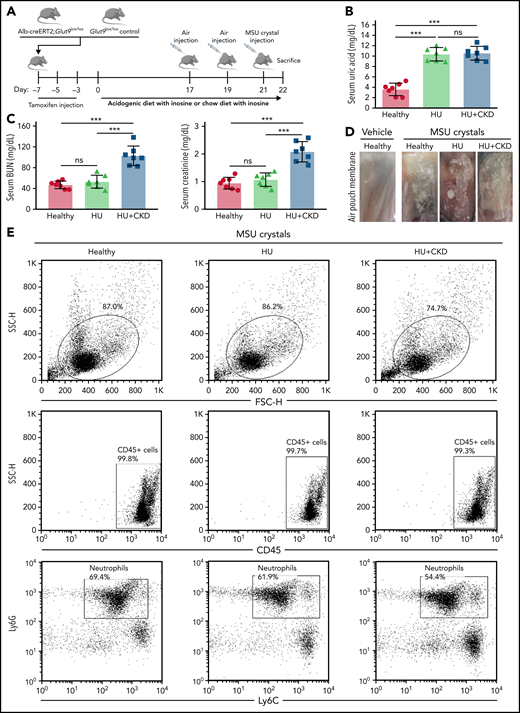
To investigate whether HU might have immunoregulatory effects on neutrophil recruitment during acute sterile inflammation in vivo, we used a previously described transgenic mouse model of HU (Figure 1A),28 whereby Alb-creERT2;Glut9lox/lox mice were fed an inosine-rich diet. These mice developed HU in a range of 9 to 12 mg/dL, similar to levels found in CKD patients, whereas Glut9lox/lox control mice displayed normal serum UA levels (Figure 1B). Feeding mice an acidogenic diet along with inosine resulted in HU plus UA nephropathy (CKD) as documented by elevated BUN and creatinine levels as well as diffuse tubular atrophy and interstitial fibrosis (Figure 1C; supplemental Figure 1A-G). At baseline, we did not observe differences in the number of CD45+ leukocytes and CD45+CD11b+Ly6G+Ly6C− neutrophils in the bone marrow, blood, and tissues between the 3 groups (supplemental Figure 2A-B). Only mice with HU plus CKD showed an increased number of CD45+ leukocytes and neutrophils in the kidney compared with healthy and HU mice (supplemental Figure 2A-B) as a result of kidney disease–related inflammation (supplemental Figure 1A-G).

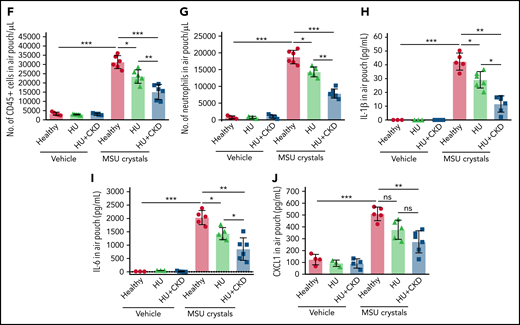
Leukocyte recruitment is impaired in sterile inflammation in hyperuricemic mice. (A) Alb-creERT2;Glut9lox/lox mice and Glut9lox/lox control mice were injected intraperitoneally with tamoxifen. Both groups were fed either an acidogenic diet enriched with inosine or a standard chow diet with inosine for 22 days. On day 21, mice received a subcutaneous injection of monosodium urate (MSU) crystals (5 mg) or vehicle into a preexisting air pouch and were euthanized 12 hours later. (B-C) Serum uric acid (B), blood urea nitrogen (BUN), and creatinine (C) levels of Glut9lox/lox mice with chow diet and inosine (healthy), Alb-creERT2;Glut9lox/lox mice with chow diet (HU), or acidogenic diet with inosine (HU plus CKD) on day 22 (n = 7 mice per group) (using 1-way ANOVA). (D) Representative images of vehicle and MSU crystals injected into the air pouch. (E-G) Gating strategy (E) and number (No.) of CD45+ leukocytes (F) and neutrophils (G) in air pouch per µL from mice with or without MSU crystals determined by flow cytometry (n = 3-6 mice per group). (H-J) Concentrations of IL-1β (H), IL-6 (I), and CXCL1 (J) measured in the air pouch fluid via ELISA (n = 3-6 mice per group). Data are mean plus or minus SD. *P < .05, **P < .01, ***P < .001. ns, not significant by 1-way ANOVA; ELISA, enzyme-linked immunoassay.
Leukocyte recruitment is impaired in sterile inflammation in hyperuricemic mice. (A) Alb-creERT2;Glut9lox/lox mice and Glut9lox/lox control mice were injected intraperitoneally with tamoxifen. Both groups were fed either an acidogenic diet enriched with inosine or a standard chow diet with inosine for 22 days. On day 21, mice received a subcutaneous injection of monosodium urate (MSU) crystals (5 mg) or vehicle into a preexisting air pouch and were euthanized 12 hours later. (B-C) Serum uric acid (B), blood urea nitrogen (BUN), and creatinine (C) levels of Glut9lox/lox mice with chow diet and inosine (healthy), Alb-creERT2;Glut9lox/lox mice with chow diet (HU), or acidogenic diet with inosine (HU plus CKD) on day 22 (n = 7 mice per group) (using 1-way ANOVA). (D) Representative images of vehicle and MSU crystals injected into the air pouch. (E-G) Gating strategy (E) and number (No.) of CD45+ leukocytes (F) and neutrophils (G) in air pouch per µL from mice with or without MSU crystals determined by flow cytometry (n = 3-6 mice per group). (H-J) Concentrations of IL-1β (H), IL-6 (I), and CXCL1 (J) measured in the air pouch fluid via ELISA (n = 3-6 mice per group). Data are mean plus or minus SD. *P < .05, **P < .01, ***P < .001. ns, not significant by 1-way ANOVA; ELISA, enzyme-linked immunoassay.
To investigate the effect of HU in acute sterile inflammation, we injected MSU crystals or vehicle into dorsal subcutaneous air pouches in all 3 groups of mice (Figure 1D). Twelve hours later, flow cytometry of air pouch lavage fluids revealed a significant increase in infiltrating CD45+ leukocytes and CD45+CD11b+Ly6G+Ly6C− neutrophils in mice injected with MSU crystals (Figure 1E-G). However, mice with HU displayed significantly fewer neutrophils in pouch fluids, an effect that was even more pronounced in mice with concomitant UA nephropathy (CKD) (Figure 1E-G). The impaired migratory potential of neutrophils was associated with a downregulation of the β2 integrin MAC-1 in blood neutrophils from HU and HU plus CKD mice as compared with the control group (supplemental Figure 2C). In addition, pouch fluid levels of IL-1β, IL-6, and CXCL-1 were reduced in HU and HU plus CKD mice accordingly (Figure 1H-J). These mice were also injected with DAMP/PAMP supernatants derived from L929 cells infected with the mouse hepatitis virus, a murine coronavirus, into air pouches and showed similar results (supplemental Figure 3). Thus, HU with and without kidney dysfunction attenuates sterile inflammation by suppressing neutrophil recruitment.
To test for the effect of kidney disease alone, MSU crystals were also injected into air pouches in mice with oxalate-rich diet–induced CKD lacking respective HU (supplemental Figure 4A-D). We found that the number of infiltrating CD45+ leukocytes and neutrophils into the air pouch was significantly reduced in CKD mice as compared with control mice (supplemental Figure 4E-F), whereas the levels of the proinflammatory cytokines IL-1β and IL-6 (supplemental Figure 4G-H) did not change between the groups after MSU crystal injection.
In addition, using a model of acute ischemic organ injury (supplemental Figure 5A), we found significantly less tubular injury, lower serum IL-6 concentrations, and reduced numbers of kidney CD45+ leukocytes and neutrophils in HU mice (HU plus ischemia reperfusion injury) compared with the control group (healthy plus ischemia reperfusion injury) at day 1 after ischemia reperfusion injury (supplemental Figure 5B-G).
Taken together, the data shows that HU with or without CKD attenuates MSU crystal- and coronavirus-related DAMP/PAMP-induced sterile inflammation by suppressing leukocyte, in particular, neutrophil recruitment, whereas CKD alone had a selective effect on neutrophil recruitment but not on the inflammatory response per se.
Hyperuricemia contributes to kidney dysfunction–related suppression of sterile inflammation in mice
To investigate the putative contribution of HU on the effects seen during acute sterile inflammation, we treated HU plus CKD mice with vehicle or rasburicase, a Food and Drug Administration–approved and commercially available urate-depleting therapy (UDT) that specifically targets UA to lower serum UA levels in humans29 and mice.30 Rasburicase treatment significantly decreased serum sUA levels in HU plus CKD mice without affecting BUN levels (Figure 2A-B) or MSU crystal deposition in the air pouch itself (Figure 2D). Rasburicase did not affect serum hydrogen peroxide levels, a potential biomarker of oxidative stress (Figure 2C). In addition, flow cytometry analysis revealed that correcting HU with rasburicase partially recovered the number of CD45+ leukocytes and neutrophils migrating into the air pouch filled with MSU crystals (Figure 2E-F) as well as the pouch fluid concentrations of IL-1β, IL-6, and CXCL-1 (Figure 2G-I) compared with vehicle-treated HU plus CKD and healthy mice. Thus, HU acts as an endogenous suppressor of sterile inflammation in kidney dysfunction.

Rasburicase treatment reverses the impaired leukocyte recruitment and inflammation in mice with hyperuricemia and kidney dysfunction. Alb-creERT2;Glut9lox/lox and Glut9lox/lox control mice were injected with tamoxifen and placed on an acidogenic diet with inosine (HU plus CKD) or a chow diet with inosine (healthy) for 22 days. HU plus CKD mice received vehicle or rasburicase treatment prior to injection of vehicle (PBS) or MSU crystals (5 mg) into a preexisting air pouch and were euthanized after 12 hours. (A-B) Serum uric acid (A) and BUN (B) levels of HU plus CKD mice with rasburicase or vehicle treatment (n = 5 per group, Student t test). (C) Serum hydrogen peroxide (H2O2) levels measured in HU plus CKD mice treated with rasburicase or vehicle (n = 5 per group, Student t test). (D) Representative images of MSU crystal injection into the air pouch with or without rasburicase. (E-F) Flow cytometry analysis of infiltrating CD45+ leukocytes (E) and neutrophils (F) into the air pouch from mice with or without MSU crystals and/or rasburicase treatment (n = 4-6 per group). (G-I) Concentrations of IL-1β (G), IL-6 (H), and CXCL1 (I) from air pouch fluid measured via ELISA (n = 4-6 per group). Data are mean plus or minus SD. *P < .05, **P < .01, ***P < .001. ns, not significant by 2-way ANOVA; PBS, phosphate-buffered saline; ELISA, enzyme-linked immunoassay.
Rasburicase treatment reverses the impaired leukocyte recruitment and inflammation in mice with hyperuricemia and kidney dysfunction. Alb-creERT2;Glut9lox/lox and Glut9lox/lox control mice were injected with tamoxifen and placed on an acidogenic diet with inosine (HU plus CKD) or a chow diet with inosine (healthy) for 22 days. HU plus CKD mice received vehicle or rasburicase treatment prior to injection of vehicle (PBS) or MSU crystals (5 mg) into a preexisting air pouch and were euthanized after 12 hours. (A-B) Serum uric acid (A) and BUN (B) levels of HU plus CKD mice with rasburicase or vehicle treatment (n = 5 per group, Student t test). (C) Serum hydrogen peroxide (H2O2) levels measured in HU plus CKD mice treated with rasburicase or vehicle (n = 5 per group, Student t test). (D) Representative images of MSU crystal injection into the air pouch with or without rasburicase. (E-F) Flow cytometry analysis of infiltrating CD45+ leukocytes (E) and neutrophils (F) into the air pouch from mice with or without MSU crystals and/or rasburicase treatment (n = 4-6 per group). (G-I) Concentrations of IL-1β (G), IL-6 (H), and CXCL1 (I) from air pouch fluid measured via ELISA (n = 4-6 per group). Data are mean plus or minus SD. *P < .05, **P < .01, ***P < .001. ns, not significant by 2-way ANOVA; PBS, phosphate-buffered saline; ELISA, enzyme-linked immunoassay.
Hyperuricemia attenuates neutrophil adhesion and extravasation in mice
To dissect the effect of HU on the different steps of the leukocyte recruitment cascade, we performed intravital microscopy of postcapillary venules in the cremaster muscle of mice (Figure 3A) and investigated leukocyte rolling, leukocyte rolling velocity, leukocyte adhesion efficiency, and leukocyte extravasation into inflamed tissue.31,32 Importantly, hemodynamic parameters (vessel diameter, centerline velocity, and shear rates) were identical in all 4 groups of mice (supplemental Table 2). Following crystal injection, the adhesion efficiency was significantly reduced in vessels of HU mice as compared with control mice, which was even more pronounced in crystal injected HU plus CKD mice (Figure 3B), indicating that HU impairs β2 integrin–dependent firm arrest of leukocytes. A representative video is available in the supplements (supplemental Video 1). HU significantly increased the rolling velocity of interacting leukocytes (Figure 3C-D), suggesting reduced activation of β2 integrins, which is critical for slow rolling velocities. Of note, overall leukocyte rolling was not affected (Figure 3E). In line with the reduced intravasal adhesion of leukocytes in HU and HU plus CKD mice, quantifying perivascular leukocyte subsets in whole-mount Giemsa-stained cremaster muscles mirrored these effects within the inflamed tissue (Figure 3F). Differential cell counts displayed reduced numbers of perivascular neutrophils and mononuclear cells in HU mice (Figure 3F). Moreover, treating HU plus CKD mice with rasburicase restored the migratory potential of neutrophils as indicated by increased adhesion efficiency (Figure 3G) and number of perivascular leukocytes (Figure 3J) but decreased rolling velocity (Figure 3H-I). Hemodynamic parameters (vessel diameter, centerline velocity, and shear rates) were similar in vehicle- and rasburicase-treated HU plus CKD mice (supplemental Table 3). Taken together, 2 in vivo models consistently show that HU decreases neutrophil adhesion and extravasation toward the site of sterile inflammation.

Decreased neutrophil adhesion and extravasation in crystal-stimulated cremaster muscle venules in hyperuricemic mice. (A) Alb-creERT2;Glut9lox/lox and Glut9lox/lox control mice were injected with tamoxifen and placed on a chow or acidogenic diet with inosine (n = 5-7 mice per group). HU plus CKD mice received vehicle or rasburicase treatment. After 22 days, mice were injected intrascrotally with MSU crystals (0.5 mg in 200 µL NaCl per mouse) or vehicle (200 µL NaCl per mouse) 4 hours prior to intravital microscopy. (B) Quantification of adhesion efficiency (n = 5-7 mice per group). (C-D) Cumulative distribution (C) and mean leukocyte rolling velocity (µm per second) (D). (E) Quantification of leukocyte rolling depicted as rolling flux fraction. (F) The total number of perivascular neutrophils and mononuclear cells per mm2 after MSU crystal injection (n = 5 mice per group). (G) Quantification of the adhesion efficiency (n = 5 mice per group). (H-I) Cumulative frequency over leukocyte rolling velocity (H) and mean of rolling velocity (µm per second) (I). (J) The total number of perivascular neutrophils and mononuclear cells per mm2 after MSU crystal injection in vehicle- and rasburicase-treated HU plus CKD mice (n = 4-5 mice per group). Data are mean plus or minus SD. *P < .05, **P < .01, ***P < .001. ns, not significant by Student t test or 1- or 2-way ANOVA.
Decreased neutrophil adhesion and extravasation in crystal-stimulated cremaster muscle venules in hyperuricemic mice. (A) Alb-creERT2;Glut9lox/lox and Glut9lox/lox control mice were injected with tamoxifen and placed on a chow or acidogenic diet with inosine (n = 5-7 mice per group). HU plus CKD mice received vehicle or rasburicase treatment. After 22 days, mice were injected intrascrotally with MSU crystals (0.5 mg in 200 µL NaCl per mouse) or vehicle (200 µL NaCl per mouse) 4 hours prior to intravital microscopy. (B) Quantification of adhesion efficiency (n = 5-7 mice per group). (C-D) Cumulative distribution (C) and mean leukocyte rolling velocity (µm per second) (D). (E) Quantification of leukocyte rolling depicted as rolling flux fraction. (F) The total number of perivascular neutrophils and mononuclear cells per mm2 after MSU crystal injection (n = 5 mice per group). (G) Quantification of the adhesion efficiency (n = 5 mice per group). (H-I) Cumulative frequency over leukocyte rolling velocity (H) and mean of rolling velocity (µm per second) (I). (J) The total number of perivascular neutrophils and mononuclear cells per mm2 after MSU crystal injection in vehicle- and rasburicase-treated HU plus CKD mice (n = 4-5 mice per group). Data are mean plus or minus SD. *P < .05, **P < .01, ***P < .001. ns, not significant by Student t test or 1- or 2-way ANOVA.
Soluble uric acid inhibits β2 integrin activation and prevents efficient neutrophil migration in vitro
To elucidate the underlying molecular mechanisms of how HU could affect neutrophil rolling velocity, neutrophil adhesion, and extravasation in vivo, we studied the impact of sUA on β2 integrin activation in blood neutrophils isolated from healthy individuals in vitro (Figure 4A). β2 integrin exists in 3 different conformation/activation states: a closed, inactive, and bent conformation; an intermediate extended active conformation; and an open, fully extended confirmation,33 as illustrated in Figure 4B. Using the β2 integrin activation marker mAB24, which only recognizes the open, fully extended confirmation in human neutrophils, we investigated the effects of sUA on CXCL8-induced β2-integrin activation. Human neutrophils were stimulated for 30 minutes with 10 mg/dL sUA or vehicle, followed by 5 minutes of CXCL8 stimulation. Stimulation of control neutrophils with CXCL8 induced β2 integrin activation, as indicated by increased mAB24 binding and MFI compared with medium (Figure 4C-D). In contrast, pretreatment of neutrophils with sUA reduced CXCL8-induced β2 integrin activation, suggesting a role for sUA in chemokine-induced integrin activation (inside-out signaling) (Figure 4D). In addition, total surface expression of αL (LFA-1) and αM (MAC-1) subunits of β2 integrin was quantified by flow cytometry. Although activation of healthy human neutrophils with CXCL8 alone did not affect total LFA-1 expression, preincubation with sUA significantly reduced the total amount of LFA-1 in unstimulated and CXCL8-stimulated neutrophils (Figure 4E). In contrast to LFA-1, MAC-1, which is stored in intracellular pools,34 was rapidly translocated to the cellular surface upon CXCL8 stimulation. However, in the presence of sUA, CXCL8-induced MAC-1 expression was reduced (Figure 4F). Similar effects of sUA were observed for mAB24 binding, as well as for expression of LFA-1 and MAC-1 in phorbol myristate acetate–stimulated healthy human neutrophils (supplemental Figure 6). In addition, sUA also decreased total surface expression of the chemokine receptor CXCR1, whereas the expression of the chemokine receptors CXCR2 and FPR1 remained unaffected in unstimulated or CXCL8- and fMLP-stimulated neutrophils (supplemental Figure 7A-C).

Soluble uric acid affects β2 integrin activation and prevents human neutrophil migration and phagocytosis in vitro. (A) Human neutrophils were isolated from healthy individuals and preincubated with or without 10 mg/dL sUA for 30 minutes prior to stimulation with human CXCL8 or SARS-CoV2–derived DAMP/PAMP supernatant. (B) Schematic of the 3 β2 integrin conformations that reflect the different affinity stages: (1) the bent form, inactive, (2) the extended form with a closed ligand-binding head of intermediate affinity, and (3) the extended form enabling the ligand-binding for mAB24 with high affinity (adopted from Evans et al).82 (C-D) mAB24 binding illustrated as a histogram (C) and the expression levels of mAB24 (D) determined by flow cytometry (n = 6-7 per group). (E-F) The expression levels of LFA-1 (E) and MAC-1 (F) shown as mean fluorescence intensity (MFI) relative to isotype control (n = 6-7 per group) were quantified by flow cytometry. (G-H) Transwell migration assays were carried out, and the total number of neutrophils that migrated toward the chemoattractants human CXCL8 and IL-1β (G) and fMLP (H) was determined after 3 hours by flow cytometry (n = 6-12 per group). (I-J) The expression levels of LFA-1 (I) and MAC-1 (J) shown as MFI relative to isotype control (n = 4 per group) were quantified by flow cytometry. (K) Transwell migration assays were carried out, and the total number of neutrophils that migrated toward the SARS-CoV2 supernatant (20%) was determined after 3 hours by flow cytometry (n = 4 per group). (L) Percentage (%) of phagocytosed IgG FITC-latex beads in untreated or CXCL8-stimulated neutrophils with or without sUA preincubation was determined by flow cytometry (n = 4 per group). Data are mean plus or minus SD. *P < .05, **P < .01, ***P < .001. ns, not significant by 2-way ANOVA; Ctrl, control; IgG, immunoglobulin G.
Soluble uric acid affects β2 integrin activation and prevents human neutrophil migration and phagocytosis in vitro. (A) Human neutrophils were isolated from healthy individuals and preincubated with or without 10 mg/dL sUA for 30 minutes prior to stimulation with human CXCL8 or SARS-CoV2–derived DAMP/PAMP supernatant. (B) Schematic of the 3 β2 integrin conformations that reflect the different affinity stages: (1) the bent form, inactive, (2) the extended form with a closed ligand-binding head of intermediate affinity, and (3) the extended form enabling the ligand-binding for mAB24 with high affinity (adopted from Evans et al).82 (C-D) mAB24 binding illustrated as a histogram (C) and the expression levels of mAB24 (D) determined by flow cytometry (n = 6-7 per group). (E-F) The expression levels of LFA-1 (E) and MAC-1 (F) shown as mean fluorescence intensity (MFI) relative to isotype control (n = 6-7 per group) were quantified by flow cytometry. (G-H) Transwell migration assays were carried out, and the total number of neutrophils that migrated toward the chemoattractants human CXCL8 and IL-1β (G) and fMLP (H) was determined after 3 hours by flow cytometry (n = 6-12 per group). (I-J) The expression levels of LFA-1 (I) and MAC-1 (J) shown as MFI relative to isotype control (n = 4 per group) were quantified by flow cytometry. (K) Transwell migration assays were carried out, and the total number of neutrophils that migrated toward the SARS-CoV2 supernatant (20%) was determined after 3 hours by flow cytometry (n = 4 per group). (L) Percentage (%) of phagocytosed IgG FITC-latex beads in untreated or CXCL8-stimulated neutrophils with or without sUA preincubation was determined by flow cytometry (n = 4 per group). Data are mean plus or minus SD. *P < .05, **P < .01, ***P < .001. ns, not significant by 2-way ANOVA; Ctrl, control; IgG, immunoglobulin G.
In addition, we stimulated human neutrophils with DAMP/PAMP containing supernatants derived from a human hepatoma Huh7 cell line infected with SARS-CoV2. This led to a significant increase in LFA-1 and MAC-1 expression in SARS-CoV2 supernatant-treated neutrophils (SARS-CoV2 SN) compared with control neutrophils (Ctrl-SN) (Figure 4I-J). In contrast, supernatant-treated neutrophils preincubated with sUA demonstrated a significantly reduced LFA-1 and MAC-1 surface expression (SARS-CoV2-SN) (Figure 4I-J). Expression of chemokine receptor CXCR1 was also decreased in supernatant-treated preincubated with sUA, whereas the expression of CXCR2 and FPR1 remained unaffected (supplemental Figure 7D-F). Of note, similar to medium alone or control supernatants, incubation of neutrophils with sUA or SARS-CoV2 supernatants did not induce cell death (supplemental Figure 8). These results point toward a negative regulatory role of sUA on β2 integrin activation and translocation from intracellular pools to the surface.
Next, we tested the effect of sUA on neutrophils sensing chemokine gradients using transwell migration assays. We preincubated neutrophils from healthy individuals with vehicle (control) or 10 mg/dL sUA for 30 minutes and allowed neutrophils to migrate across the transwell filter in the presence or absence of different chemokines, cytokines, bacterial peptides, and coronavirus-related DAMP/PAMP supernatants. The presence of sUA attenuated the migratory potential of neutrophils toward CXCL8, IL-1β (Figure 4G), the bacterial peptide fMLP (Figure 4H), and SARS-CoV2–related DAMP/PAMP supernatant (Figure 4K). This is consistent with a reduced expression of CXCR1 (supplemental Figure 7A-D), a receptor important for neutrophil chemotaxis.35 In addition, the phagocytic capability of neutrophils to take up IgG FITC-latex beads were also impaired in the presence of sUA (Figure 4L), indicating that sUA affects the migratory and phagocytic capability of neutrophils.
Soluble uric acid affects β2 integrin internalization and recycling in human neutrophils
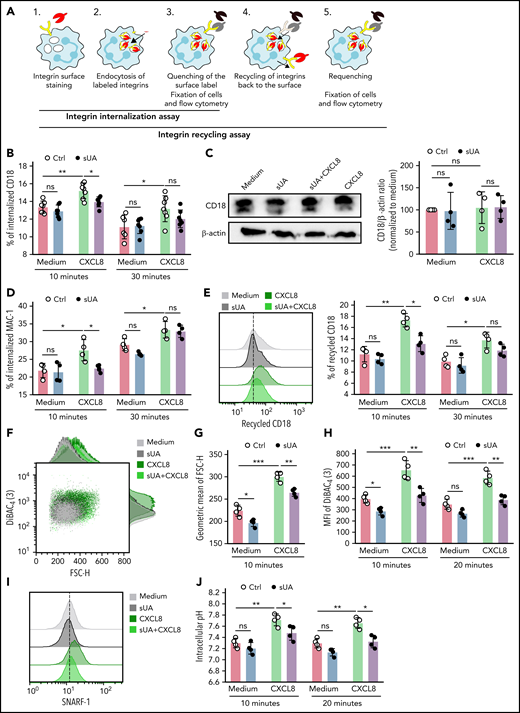
Integrins undergo constant endo-/exocytic traffic in neutrophils,36 whereby cell surface integrins are internalized/endocytosed and recycled back to the cell membrane, a mechanism important for cell migration. We speculated that sUA would affect neutrophil migration by interfering with β2 integrin internalization as a central regulator of the migration process. To test for this possibility, we preincubated human blood neutrophils from healthy subjects with or without 10 mg/dL sUA prior to stimulation with human CXCL8 for 30 minutes and performed integrin internalization assays (Figure 5A).37 Flow cytometric analysis revealed that the percentage of internalized CD18 significantly increased in CXCL8-activated neutrophils as compared with medium and sUA after 10 and 30 minutes (Figure 5B). However, sUA diminished CD18 internalization in CXCL8-activated neutrophils, as indicated by a reduced percentage of internalized CD18 (Figure 5B). Western immunoblotting confirmed that sUA did not change the total amount of CD18 in CXCL8-activated neutrophils (Figure 5C). Soluble UA also decreased the percentage of internalized MAC-1 (Figure 5D) and diminished CD18 recycling in CXCL8-activated neutrophils after 10 minutes (Figure 5E).

Soluble uric acid affects β2 integrin internalization and recycling in human neutrophils. (A) Schematic illustrating the process of integrin internalization and recycling in neutrophils (detailed information is described in supplemental Methods). (B) Human neutrophils were isolated from healthy individuals and preincubated with or without 10 mg/dL sUA for 30 minutes prior to stimulation with CXCL8. The integrin internalization assay for anti-human CD18 in CXCL8-activated neutrophils was performed using flow cytometry. The percentage (%) of internalized CD18 in CXCL8-activated neutrophils was determined after 10 and 30 minutes (n = 7 per group). (C) Immunoblotting for CD18 and β-actin was performed, and the protein CD18/β-actin ratio to medium quantified (n = 4 per group). (D) The integrin internalization assay was performed and the percentage of internalized MAC-1 after 10 and 30 minutes determined by flow cytometry (n = 4 per group). (E) The integrin recycling assay was performed and the percentage of recycled CD18 after 10 and 30 minutes determined by flow cytometry (n = 4 per group). (F-H) Cytoskeletal dynamics were determined in CXCL8-activated neutrophils (n = 4 per group) via flow cytometry using the forward scatter (FSC-H) for quantifying cellular size (F-G) and the dye DiBAC4(3) for quantification of membrane potential (as MFI) (H). (I-J) Intracellular pH was determined as MFI of the dye SNARF-1 (MFI) (I) by flow cytometry and the intracellular pH calculated (J) (n = 4 per group). Data are mean plus or minus SD. *P < .05, **P < .01. Ctrl, control; ns, not significant by 2-way ANOVA.
Soluble uric acid affects β2 integrin internalization and recycling in human neutrophils. (A) Schematic illustrating the process of integrin internalization and recycling in neutrophils (detailed information is described in supplemental Methods). (B) Human neutrophils were isolated from healthy individuals and preincubated with or without 10 mg/dL sUA for 30 minutes prior to stimulation with CXCL8. The integrin internalization assay for anti-human CD18 in CXCL8-activated neutrophils was performed using flow cytometry. The percentage (%) of internalized CD18 in CXCL8-activated neutrophils was determined after 10 and 30 minutes (n = 7 per group). (C) Immunoblotting for CD18 and β-actin was performed, and the protein CD18/β-actin ratio to medium quantified (n = 4 per group). (D) The integrin internalization assay was performed and the percentage of internalized MAC-1 after 10 and 30 minutes determined by flow cytometry (n = 4 per group). (E) The integrin recycling assay was performed and the percentage of recycled CD18 after 10 and 30 minutes determined by flow cytometry (n = 4 per group). (F-H) Cytoskeletal dynamics were determined in CXCL8-activated neutrophils (n = 4 per group) via flow cytometry using the forward scatter (FSC-H) for quantifying cellular size (F-G) and the dye DiBAC4(3) for quantification of membrane potential (as MFI) (H). (I-J) Intracellular pH was determined as MFI of the dye SNARF-1 (MFI) (I) by flow cytometry and the intracellular pH calculated (J) (n = 4 per group). Data are mean plus or minus SD. *P < .05, **P < .01. Ctrl, control; ns, not significant by 2-way ANOVA.
Moreover, reports suggest that neutrophil migration is associated with physiological changes in cytoskeletal dynamics and intracellular pH.38,39 Our flow cytometric analysis revealed that neutrophil size and membrane potential increased in CXCL8-activated neutrophils compared with unstimulated neutrophils, as indicated by an increase in geometric mean of forward scatter and MFI of the membrane potential dye DiBAC4(3) (Figure 5F-H). However, sUA diminished both neutrophil size and plasma membrane potential in CXCL8-activated neutrophils to values in unstimulated neutrophils (Figure 5F-H). Flow chamber assays confirmed reduced LFA-1 surface expression upon sUA treatment in murine neutrophils (supplemental Figure 9A-B). In addition, F-actin polymerization under flow was reduced in sUA-treated compared with control neutrophils, reflecting reduced β2 integrin outside-in signaling (supplemental Figure 9A-C). Furthermore, sUA also affected intracellular pH in CXCL8-activated neutrophils, as indicated by a downregulation of the cell-permeant pH indicator SNARF-1 (Figure 5I-J).
Taken together, these data show that sUA impairs β2 integrin activation and internalization/recycling. This is accompanied by changes in intracellular pH and cytoskeletal dynamics, suggesting a profound effect of intracellular sUA on cell homeostasis and function.
The immunomodulatory effect of soluble uric acid in human neutrophils involves intracellular uptake via urate transporters
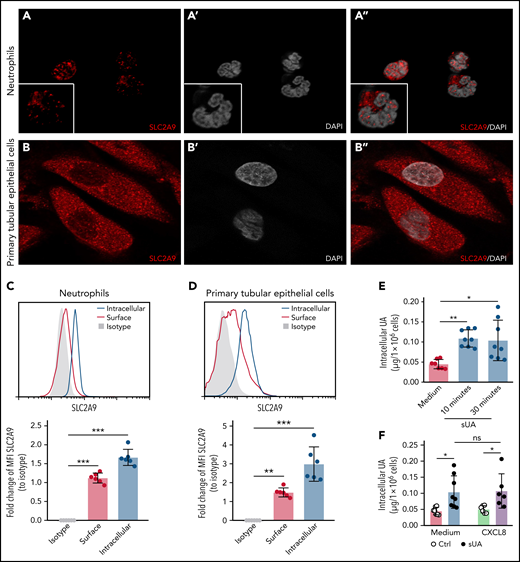
Next, we speculated that the effect of sUA on integrin recycling and function in neutrophils requires intracellular uptake of sUA via urate transporters.11 RNA sequencing data revealed that healthy human blood neutrophils mainly express the urate transporter SLC2A9 (supplemental Figure 10A)40 as compared with primary human tubular epithelial cells (TEC) that express all known urate transporters41 (supplemental Figure 10B). Using reverse transcription polymerase chain reaction analysis, we found human blood neutrophils to express the urate reabsorption transporters SLC2A9 and SLC22A13 as well as the urate excretion transporter ABCC4 (supplemental Figure 10C). Fluorescence microscopy confirmed that human neutrophils express the urate transporter SLC2A9 (Figure 6A-A"), with primary TECs cells used as positive control (Figure 6B-B"). Flow cytometry analysis confirmed that neutrophils (Figure 6C) as well as primary TECs (Figure 6D) express SLC2A9 intracellularly and on the cell surface.
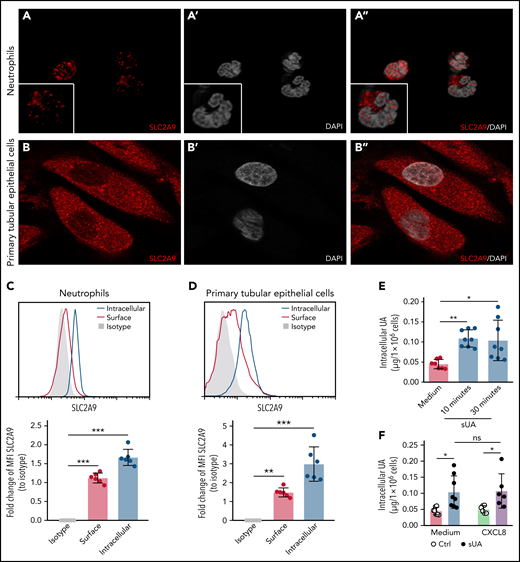
Intracellular uptake of uric acid via the urate transporter SLC2A9 in neutrophils. (A-B) Human neutrophils from healthy subjects (A, A’, and A’’) and primary human TECs (B, B’, and B’’) were stained with the anti-human SLC2A9 antibody (red) and DAPI (white, nuclei), and confocal microscopy was performed. (C-D) MFI of intracellular and surface SLC2A9 expression in human neutrophils (C) and TECs (D) compared with isotype control was determined by flow cytometry (n = 3 per group). (E) Neutrophils from healthy subjects were cultured with or without 10 mg/dL sUA for 10 and 30 minutes and the intracellular UA levels determined using an UA assay kit (n = 7-8 per group). (F) Intracellular UA levels from CXCL8-activated neutrophils in the presence or absence of 10 mg/dL sUA determined by UA assay kit (n = 6-8 per group). Data are mean plus or minus SD. *P < .05, **P < .01, ***P < .001. ns, not significant by 1-way ANOVA; DAPI, 4′,6-diamidino-2-phenylindole.
Intracellular uptake of uric acid via the urate transporter SLC2A9 in neutrophils. (A-B) Human neutrophils from healthy subjects (A, A’, and A’’) and primary human TECs (B, B’, and B’’) were stained with the anti-human SLC2A9 antibody (red) and DAPI (white, nuclei), and confocal microscopy was performed. (C-D) MFI of intracellular and surface SLC2A9 expression in human neutrophils (C) and TECs (D) compared with isotype control was determined by flow cytometry (n = 3 per group). (E) Neutrophils from healthy subjects were cultured with or without 10 mg/dL sUA for 10 and 30 minutes and the intracellular UA levels determined using an UA assay kit (n = 7-8 per group). (F) Intracellular UA levels from CXCL8-activated neutrophils in the presence or absence of 10 mg/dL sUA determined by UA assay kit (n = 6-8 per group). Data are mean plus or minus SD. *P < .05, **P < .01, ***P < .001. ns, not significant by 1-way ANOVA; DAPI, 4′,6-diamidino-2-phenylindole.
When exposing neutrophils to sUA, the intracellular levels of UA increased within 10 minutes (Figure 6E), so we speculated on a urate transporter–mediated uptake. Stimulation of sUA-treated neutrophils with human CXCL8 did not alter SLC2A9 messenger RNA expression (supplemental Figure 10D) or the amount of intracellular UA levels (Figure 6F) as compared with neutrophils not exposed to CXCL8. We conclude that neutrophils require urate transporters for the intracellular uptake of sUA, a mechanism impairing β2 integrin activity, reducing β2 integrin recycling, and compromising neutrophil chemotaxis.
Neutrophil migration but not extracellular trap release is impaired in hyperuricemic patients
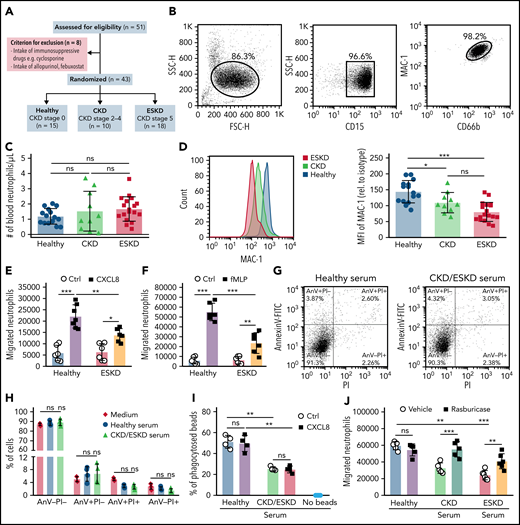
Finally, to validate the impact of HU on the functional properties of neutrophils also in hyperuricemic patients, we isolated blood neutrophils from patients with different stages of kidney dysfunction including 10 patients with CKD stage G2-4 without dialysis (CKD; male/female, 6/4; mean age, 57.1 ± 5.9 years) and 18 patients with CKD stage G5D on hemodialysis (ESKD; male/female, 11/7; mean age, 57.3 ± 3.7 years) as well as 15 healthy subjects (CKD stage 0; male/female, 7/8; mean age, 44.5 ± 4.8 years) (Figure 7A; Tables 1 and 2). Blood neutrophils (CD15+CD66b+) had a purity of 86.3% after isolation (Figure 7B). The total number of blood neutrophils was similar between the 3 groups (Figure 7C). Interestingly, neutrophils from CKD and ESKD patients revealed a significantly lower surface expression of MAC-1 compared with neutrophils from healthy subjects as determined by flow cytometry (Figure 7D). Next, we quantified the ability of neutrophils to sense chemokine gradients by performing transwell migration assays as before. In line with our data obtained with murine cells, neutrophils from patients with ESKD migrated in significantly lower numbers along a gradient of human CXCL8 and fMLP as compared with neutrophils isolated from healthy individuals (Figure 7E-F). In addition, we tested whether sUA in the serum contributes to the intrinsic dysfunction of neutrophils from CKD/ESKD patients vs healthy subjects. Therefore, we isolated neutrophils from healthy subjects and preincubated the cells with serum from either CKD and ESKD patients or healthy individuals. These sera contained sUA levels as follows: CKD (8.93 ± 0.46 mg/dL), ESKD (10.25 ± 0.55 mg/dL), and healthy (4.27 ± 0.26 mg/dL). Of note, incubation of human neutrophils with CKD/ESKD or healthy serum did not induce neutrophil cell death (Figure 7G-H). Serum from CKD and ESKD patients, however, decreased the ability of neutrophils from healthy donors to phagocytose IgG FITC-latex beads (Figure 7I) and to migrate toward fMLP as compared with neutrophils incubated with serum from healthy individuals. This effect was reversed by depleting sUA from the serum with rasburicase before neutrophil incubation (Figure 7J). No difference in activated neutrophils to release NETs42 was observed between hyperuricemic ESKD patients and healthy subjects (supplemental Figure 11). These data consistently indicate that kidney dysfunction–related HU impairs the migratory and phagocytic ability of neutrophils without affecting NET release.
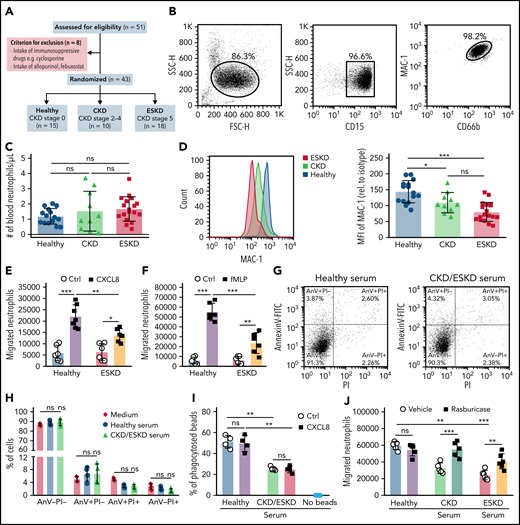
Soluble uric acid impairs neutrophil migration in patients with kidney dysfunction. (A) Schematic of study design. Out of 51 individuals, 8 patients were excluded due to the intake of immunosuppressive drugs or xanthine oxidase inhibitors. Of the remaining, 10 patients represented with CKD (CKD stage G2-4), 18 patients with ESKD that were on hemodialysis (CKD stage G5), and 15 healthy individuals without any renal pathologies (healthy, CKD stage 0). (B) Human neutrophils were isolated and identified as CD15+CD66b+ by flow cytometry (with gating strategy) with a purity of ∼99%. (C) Number of human neutrophils that were isolated from patients with kidney dysfunction (CKD and ESKD) as well as healthy individuals. (D) Expression (MFI) of MAC-1 relative to isotype determined by flow cytometry (healthy, n = 15; CKD, n = 10; ESKD, n = 18; 1-way ANOVA). (E-F) The total number of neutrophils isolated from healthy individuals and ESKD patients that migrated toward the chemoattractants human CXCL8 (E) and fMLP (F) (healthy, n = 6-7; ESKD, n = 6; 2-way ANOVA) was determined by flow cytometry after 3 hours. (G-H) Neutrophils were incubated with medium and serum from healthy individuals or CKD/ESKD patients and AnnexinV (AnV)-PI staining performed to quantify cell death by flow cytometry. Representative images of dot plots (G) and the percentage (%) of live (AnV-PI−), apoptotic (AnV+PI−), late apoptotic/early necrotic (AnV+PI+), and necrotic (AnV-PI+) neutrophils (H). (I) Percentage (%) of phagocytosed IgG FITC-latex beads in untreated or CXCL8-stimulated neutrophils with or without preincubation of sera from healthy individuals or CKD/ESKD patients was determined by flow cytometry (n = 4 per group; 2-way ANOVA). (J) Healthy neutrophils were incubated with serum from healthy individuals or patients with kidney dysfunction in the absence or presence of rasburicase, and the total number of neutrophils that migrated toward fMLP was determined by flow cytometry after 3 hours (n = 5-6 per group; 2-way ANOVA). Data are mean plus or minus SD. *P < .05, **P < .01, ***P < .001. ns, not significant; Ctrl, control.
Soluble uric acid impairs neutrophil migration in patients with kidney dysfunction. (A) Schematic of study design. Out of 51 individuals, 8 patients were excluded due to the intake of immunosuppressive drugs or xanthine oxidase inhibitors. Of the remaining, 10 patients represented with CKD (CKD stage G2-4), 18 patients with ESKD that were on hemodialysis (CKD stage G5), and 15 healthy individuals without any renal pathologies (healthy, CKD stage 0). (B) Human neutrophils were isolated and identified as CD15+CD66b+ by flow cytometry (with gating strategy) with a purity of ∼99%. (C) Number of human neutrophils that were isolated from patients with kidney dysfunction (CKD and ESKD) as well as healthy individuals. (D) Expression (MFI) of MAC-1 relative to isotype determined by flow cytometry (healthy, n = 15; CKD, n = 10; ESKD, n = 18; 1-way ANOVA). (E-F) The total number of neutrophils isolated from healthy individuals and ESKD patients that migrated toward the chemoattractants human CXCL8 (E) and fMLP (F) (healthy, n = 6-7; ESKD, n = 6; 2-way ANOVA) was determined by flow cytometry after 3 hours. (G-H) Neutrophils were incubated with medium and serum from healthy individuals or CKD/ESKD patients and AnnexinV (AnV)-PI staining performed to quantify cell death by flow cytometry. Representative images of dot plots (G) and the percentage (%) of live (AnV-PI−), apoptotic (AnV+PI−), late apoptotic/early necrotic (AnV+PI+), and necrotic (AnV-PI+) neutrophils (H). (I) Percentage (%) of phagocytosed IgG FITC-latex beads in untreated or CXCL8-stimulated neutrophils with or without preincubation of sera from healthy individuals or CKD/ESKD patients was determined by flow cytometry (n = 4 per group; 2-way ANOVA). (J) Healthy neutrophils were incubated with serum from healthy individuals or patients with kidney dysfunction in the absence or presence of rasburicase, and the total number of neutrophils that migrated toward fMLP was determined by flow cytometry after 3 hours (n = 5-6 per group; 2-way ANOVA). Data are mean plus or minus SD. *P < .05, **P < .01, ***P < .001. ns, not significant; Ctrl, control.
Discussion
We had hypothesized that sUA would have immunomodulatory effects on blood neutrophils during sterile inflammation in kidney dysfunction. Indeed, our in vivo and in vitro data clearly identify that the intracellular uptake of sUA via urate transporters suppresses innate immune effector functions of neutrophils. Most interestingly, sUA regulates β2 integrin activity and internalization/recycling via regulating intracellular pH and cytoskeletal dynamics, thereby impairing neutrophils capacity to migrate and to phagocytose without affecting NET release. Thus, sUA acts as an endogenous negative regulator of innate immunity in kidney dysfunction.
Hyperuricemia is strongly linked with acute UA nephropathy, which develops most often due to tumor lysis syndrome induced by radio- or chemotherapy received for rapidly proliferating hematologic malignancies.43,44 Acute UA nephropathy occurs as a result of UA precipitation causing tubular obstruction and subsequent acute kidney injury.44 Hence, only crystalline but not sUA causes kidney injury. Support for this comes from experimental studies showing that only HU with renal UA crystal deposits but not asymptomatic HU (sUA) drives kidney dysfunction.28,45 To address the impact of HU during sterile inflammation in kidney dysfunction, we used 3 in vivo models of acute sterile inflammation based on a novel transgenic animal model of HU with or without kidney dysfunction that is finally able to reach clinically relevant levels of HU (serum UA levels of ∼10 mg/dL)28 as compared with previously published models.15,46-50 Using this innovative approach, we show that HU affects various steps of the neutrophil recruitment cascade. In particular, HU increases rolling velocity and decreases firm adhesion. This is associated with an UA-induced decrease in the expression of LFA-1 and MAC-1 leading to reduced neutrophils transmigration into the tissue and thus limits sterile inflammation. This process was partially reversible by specifically targeting UA via UDT with rasburicase. Although being a substrate for UA crystals, HU can suppress sterile inflammation. It is possible that leukemia or chemotherapy-related HU may contribute to infectious complications in this setting by promoting the acquired immunodeficiency via regulating immune cell function.
To date, limited data are available on the effects of sUA on circulating immune cell functions during sterile inflammation. Here, we report for the first time that sUA regulates β2 integrin activity and internalization/recycling in activated human neutrophils, thus impairing their migratory capability in vitro. This mechanism occurs most likely through the interference of intracellular sUA, which is taken up via urate transporters, with β2 integrin activity and internalization/recycling. Studies also reported that integrin-mediated neutrophil activation and migration are associated with changes in intracellular pH and cytoskeletal dynamics (cellular shape and actin polymerization).39,51-53 For example, inflammatory mediators including CXCL8, C5a, lipopolysaccharide, and fMLP can enhance intracellular pH (alkalization) and cytoskeletal dynamics in neutrophils, promoting rapid neutrophil recruitment to the site of inflammation,38,51,53 which is in line with our findings. Soluble UA, however, impairs these physiological processes in activated neutrophils. Consistent with these findings, the loss of MAC-1 integrin expression on neutrophils from CKD patients, similar to that observed for the CXCR1 chemokine receptor in ESKD patients,54 was associated with a diminished migratory ability of neutrophils, suggesting that HU contributes to altered integrin activity and trafficking in neutrophils from hyperuricemic CKD/ESKD patients and their persistent impaired effector function.55,56 In addition, reports have shown that leptin,57 resistin,58 modified ubiquitin,59 and granulocyte inhibitory proteins60 can impair neutrophils chemotaxis in patients with kidney dysfunction. Rossaint et al identified a role for the fibroblast growth factor FGF23, which is highly elevated in CKD/ESKD patients, to deactivate neutrophil integrins and thereby inhibiting neutrophil migration in infection and inflammation during kidney dysfunction.21,61 Several other endogenous regulators of neutrophil integrin activity and migration have been identified, including growth and differentiation factor GDF15,62 developmental endothelial locus Del-1,63 and annexin A1.64 Dysfunctions of the endothelium are also common in kidney disease, which might explain the increased HU-mediated leukocyte rolling velocity in mice due to a weak adhesive interaction between rolling leukocytes and the endothelium.65-67 Moreover, we found that sUA inhibits the phagocytic capability of human neutrophils, a function essential for host defense against infection. Phagocytic defects of neutrophils in ESKD patients have been reported,56,68 suggesting that HU might contribute to the immunodeficiency related to kidney dysfunction. Further studies are needed to investigate the effects of sUA on neutrophil cellular volume69-72 and on other integrins such as β1 integrins73 as well as the involvement of other urate transporters on neutrophil functions.74
Disturbances of immune cell functions have not only been reported for lymphocytes75-77 and neutrophils but also for human monocytes20,78 in kidney dysfunction. We recently reported that CKD-related HU inhibits the inflammatory function of monocytes to respond to danger signals, a process dependent on the intracellular uptake of sUA via SLC2A9.79 Hence, HU-driven immune dysfunction of neutrophils and monocytes might be a potential mechanism responsible for the CKD-related acquired immunodeficiency.80 Xanthine oxidase inhibitors (as urate-lowering therapy) are commonly used to lower serum UA levels and to target xanthine oxidase–related reactive oxygen species to suppress IL-1β secretion in activated macrophages, thus exhibiting anti-inflammatory effects.81 However, only UA-depleting agents like UDT specifically target UA and may allow more definite impacts in regard to the opposite roles of UA and reactive oxygen species81 in sterile inflammation and host defense.
In conclusion, we identified sUA as an endogenous negative regulator of neutrophil function. We showed that intracellular sUA modulates neutrophil recruitment by impairing efficient β2 integrin surface mobilization and activation via changes in intracellular pH and cytoskeletal dynamics in response to inflammatory danger signals, for example, during crystal-, coronavirus-, and ischemic organ injury–related inflammation, with or without kidney dysfunction. It is tempting to speculate on a role of HU also in CKD/ESKD-related secondary immunodeficiency80 and that correcting HU with UDT in kidney dysfunction may restore host defense, which remains to be addressed in future experiments.
Acknowledgments
The authors thank all patients and healthy volunteers involved in this study, the Dialysis Unit and Division of Nephrology for patient recruitment and assessment, and Frédéric Preitner and Bernhard Thorens from the University of Lausanne (Center for Integrative Genomics, Lausanne, Switzerland) for providing the Alb-creERT2,Glut9lox/lox mice, as well as Jana Mandelbaum and Anna Amifiadou for expert technical support and Yvonne Minor for animal husbandry.
This work was supported by grants from the Deutsche Forschungsgemeinschaft (DFG) STE2437/2-1, 2-2, and 4-1; the LMUexcellent Junior Researcher Fund to S.S.; the DFG AN372/14-3, 16-2, and 30-1 to H.-J.A., the DFG SFB914 project B11 to M.P., the DFG SFB914 project A5 to R.T.B; the DFG SFB914 projects B1 and Z3 to M. Sperandio; and the Bundesministerium für Bildung und Forschung RAPID 01Kl1723C/01KI2006C and the Deutsches Zentrum für Infektionsforschung TTU EI 01.806 to A.v.B.
Authorship
Contribution: S.S. and H.-J.A. designed the study concept and experiments; Q.M., C.L., Q.L., M. Sellmayr, S.S., R.I., M.P., and M.N. conducted experiments and analyzed data; P.R. provided the human renal progenitor cells; R.T.B. provided the protocol and constructive suggestions for the integrin internalization assays; A.v.B., B.v.B., R.E., and R.W. provided the mouse hepatitis virus and SARS-CoV2 supernatants, respectively; S.S., Q.M., R.I., M.P., M. Sperandio, and H.-J.A. wrote the manuscript; and all contributing authors read and revised the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Stefanie Steiger, Medizinische Klinik und Poliklinik IV, Klinikum der Universität München, Ziemssenstr. 5, 80336 München, Germany; e-mail: stefanie.steiger@med.uni-muenchen.de.
Requests for data sharing may be submitted to Stefanie Steiger (stefanie.steiger@med.uni-muenchen.de).
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.