Abstract
Integrins are transmembrane receptors that mediate cell-cell and cell-extracellular matrix adhesion. Although all integrins can undergo activation (affinity change for ligands), the degree of activation is most spectacular for integrins on blood cells. The β2 integrins are exclusively expressed on the surface of all leukocytes including neutrophils, lymphocytes, and monocytes. They are essential for many leukocyte functions and are strictly required for neutrophil arrest from rolling. The inside-out integrin activation process receives input from chemokine receptors and adhesion molecules. The integrin activation pathway involves many cytoplasmic signaling molecules such as spleen tyrosine kinase, other kinases like Bruton's tyrosine kinase, phosphoinositide 3-kinases, phospholipases, Rap1 GTPases, and the Rap1-GTP-interacting adapter molecule. These signaling events ultimately converge on talin-1 and kindlin-3, which bind to the integrin β cytoplasmic domain and induce integrin conformational changes: extension and high affinity for ligand. Here, we review recent structural and functional insights into how talin-1 and kindlin-3 enable integrin activation, with a focus on the distal signaling components that trigger β2 integrin conformational changes and leukocyte adhesion under flow.
Introduction
Leukocytes are essential for fighting bacterial, viral, and fungal infections. In vertebrates, leukocytes must be able to rapidly exit the vascular system at sites of inflammation. To leave the circulation, leukocytes in blood first roll on the wall of inflamed venules1 before they arrest in response to chemokine stimulation2-5 (Figure 1A). This process requires integrin activation, a rapid change in the conformation of leukocyte integrins, which causes a dramatic increase in their affinity for their ligands (∼10 000-fold6) exposed on endothelial cells.2,3
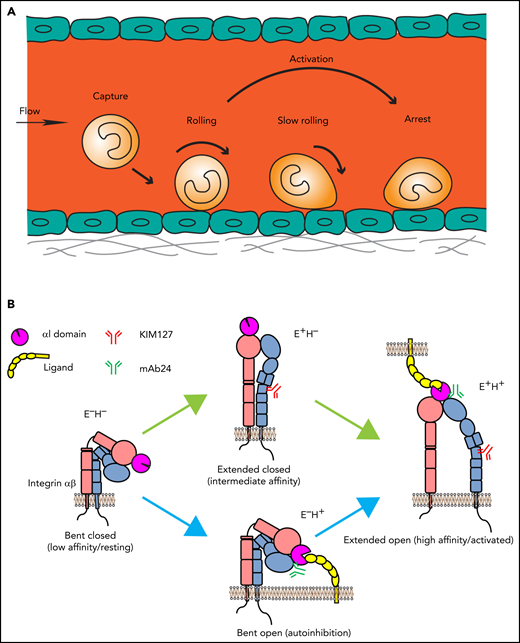
Leukocyte adhesion and integrin activation pathways. (A) Leukocyte adhesion cascade. Leukocytes circulating in the blood stream are first captured by the vessel wall, followed by rolling, slow rolling, full activation, and arrest. After arrest, leukocytes further spread and crawl along the vessel until they transmigrate the vessel wall. Rolling is mediated by selectin-ligand interactions, and arrest requires β2 integrin activation. (B) Integrin activation states and pathways. Integrins are heterodimeric proteins, consisting of an α and a β subunit. Integrin activation means the resting bent low affinity integrins acquire an H+ conformation with extended legs and an open headpiece. The open αI domain binds ligand ICAM-1 on the endothelium. Extended (E+) and H+ integrins with open αI domain conformations are reported by 2 monoclonal Abs, KIM127 and mAb24, respectively. Canonical integrin activation occurs through E+ before acquiring open αI conformation with H+. The alternative pathway starts with headpiece opening (H+) before acquiring full activation with extension and open headpiece (E+H+).
Leukocyte adhesion and integrin activation pathways. (A) Leukocyte adhesion cascade. Leukocytes circulating in the blood stream are first captured by the vessel wall, followed by rolling, slow rolling, full activation, and arrest. After arrest, leukocytes further spread and crawl along the vessel until they transmigrate the vessel wall. Rolling is mediated by selectin-ligand interactions, and arrest requires β2 integrin activation. (B) Integrin activation states and pathways. Integrins are heterodimeric proteins, consisting of an α and a β subunit. Integrin activation means the resting bent low affinity integrins acquire an H+ conformation with extended legs and an open headpiece. The open αI domain binds ligand ICAM-1 on the endothelium. Extended (E+) and H+ integrins with open αI domain conformations are reported by 2 monoclonal Abs, KIM127 and mAb24, respectively. Canonical integrin activation occurs through E+ before acquiring open αI conformation with H+. The alternative pathway starts with headpiece opening (H+) before acquiring full activation with extension and open headpiece (E+H+).
Integrins are heterodimeric αβ transmembrane receptors that anchor cells in extracellular matrix or bind counterreceptors on other cells.7 Leukocytes express at least 1 of the 4 β2 integrins.8 Circulating leukocytes within the blood stream have their β2 integrins in an off state, but when they roll along inflamed venular endothelium, their β2 integrins become partially activated,9,10 triggered by engagement of the rolling receptor P-selectin glycoprotein ligand-1 (PSGL-1).9,10 During inflammation, chemokines like interleukin 8 (IL-8) are released by macrophages and become immobilized on the endothelial surface.11 Rolling leukocytes expressing cognate receptors immediately arrest in response to IL-8.
In neutrophils, selectin- and chemokine-induced signals converge and ultimately result in full β2 integrin activation (integrin inside-out signaling).6,12,13 Integrin ectodomain extension (E+) and opening of the ligand-binding αI domain (high affinity, H+) represent the major changes that result in the fully active integrin conformation (E+H+; Figure 1B). KIM127 binds an epitope that is normally hidden in the bent knee of β2 (Figure 1B) and thus reports E+.14 mAb 24 binds a neoepitope in the β2 I-like domain that becomes accessible when the αI domain is in the H+ conformation.15 Neither KIM127 nor mAb24 affect integrin function or ligand binding and thus are activation reporters. Circulating leukocytes have their integrins in a bent (E−H−) conformation. Integrin activation occurs through the canonical pathway from bent to extended (E+H−) to fully activated (E+H+) integrins. Alternatively, the headpiece can open (E−H+) before the integrin extends (E+H+).16,17 Thus, E+ and the H+ conformation are uncoupled and can occur independently. The canonical and the alternative pathway contribute about equally to β2 integrin activation in neutrophils.16,17 Integrins in the E−H+ conformation cannot bind ligand in trans. Instead, E−H+ integrins bind ligands in cis (on the leukocyte surface), which has a net antiadhesive effect.16-18
The E+H+ β2 integrin conformation is required for leukocyte arrest from rolling. Impaired β2 integrin expression or defective regulation leads to severe pathologic phenotypes, including different types of leukocyte adhesion deficiencies (LADs), which can result in severe inflammation and even death.19-21 Patients with LAD type III exhibit normal integrin expression but show mutations in the FERMT3 gene, which encodes for the human integrin regulatory protein kindlin-3. Loss of kindlin-3 expression results in severe bleeding and recurrent infections because of defective platelet and leukocyte integrin activity.22-24
Integrin activation has been studied for decades.3,25,26 Compelling evidence has shown that talins and kindlins bind directly to the cytoplasmic tails of integrins and are strictly required for the final stages of integrin activation.25-28 Recent studies refined the structures of talin and kindlin, how they are activated, and how their binding affects the integrin extracellular domain. Signaling pathways leading to integrin activation are being unraveled. Here, we focus on structural and signaling insights into integrin activation.
Talin and kindlin
Inside-out β2 integrin activation requires the binding of talin-1 and kindlin-3 to the β2 integrin cytoplasmic tail.26 Talin-1 is expressed ubiquitously29 with high levels found in platelets30 and leukocytes. Although talin-1 was long thought to be the sole integrin activator, studies in cells and various model organisms revealed that integrin activation requires assistance by kindlins.
Talin-1 is a large cytoplasmic protein consisting of an N-terminal atypical 4.1 protein, ezrin, radixin, moesin (FERM) domain, also called the talin head with 4 subdomains (F0, F1, F2, and F3), and a C-terminal rod domain, which contains 13 consecutive helical bundles (R1-13) followed by a single helical dimerization domain (DD). The phosphotyrosine-binding motif in the F3 module binds to the membrane proximal NPxY/F of β2, which destabilizes the interactions between the α and β integrin subunits to trigger integrin activation.26 The talin rod harbors actin and vinculin binding sites; some of them become accessible when tensile forces act on talin. Talin requires activation before it can induce integrin activation, because intramolecular interactions between the talin head and rod domains preclude integrin binding.31 Electron microscopy (EM) and 3-dimensional image reconstruction studies indicate that inactive talin forms a compact dimer.32 It is likely that talin-1 prefers dimerization in cells that express high levels (like in platelets) or on local enrichment at the plasma membrane. Recent cryo-EM–based studies of full-length talin-1 have resolved the auto-inhibited conformation of the talin monomer (Figure 2A-C).33 In this conformation, charge-based interactions among the 13 rod domains form a compact globular structure with a diameter of ∼15 nm (Figure 2A-B).26 The F2 and F3 FERM subdomains interact with the R12 and R9 rod domains, respectively. The R12 domain covers the phosphatidylinositol 4,5-bisphosphate (PIP2)-binding surface of the F2 and F3 domains, thereby preventing interaction of inactive talin-1 with PIP2-enriched membrane patches. The cryo-EM structure also confirmed previous structural data that revealed interactions of the R9 domain with the F3 subdomain, which binds the β2 integrin cytoplasmic tail34 (Figure 2C). Binding of the talin F3 domain with the integrin tail and the talin R9 domain are mutually exclusive. Notably, the N-terminal F0 and F1 domains and the C-terminal dimerization domain were not resolved in the cryo-EM structure of talin-1, suggesting that they are freely accessible in autoinhibited talin. The presence of a flexible linker between the F1 and the F2 domains may allow first contacts of the F0F1 domains with cofactors such as Rap1, which are crucial for talin membrane recruitment and initiation of talin activation that ultimately leads to a stretched ∼60-nm-long open talin-1 conformation (Figure 2A). This transition is likely achieved through the unfolding of the integrin-binding FERM domain-containing head domains (THD: F0, F1, F2, and F3) and the actin-binding rod domains.
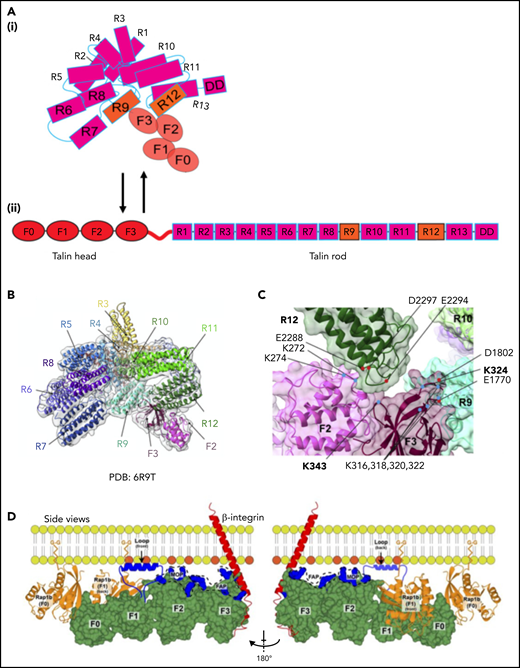
Domain organization and structure of talin-1. (A) Illustration of autoinhibited (i) and linear (ii) talin-1. Talin-1 consists of a talin head domain (THD) comprising a FERM domain (F0, F1, F2, F3, orange), and a talin rod domain (magenta). (B) Cryo-EM structure of autoinhibited human talin-1. The autoinhibited talin-1 is compactly folded with the talin-1 head interacting with the rod domains. (C) Two intramolecular interactions between the FERM domain and the rod domain, namely F3 (purple)–R9 (mint) and F2 (pink)–R12 (dark green), are shown to be critical for autoinhibition. The F0 and F1 structure is not resolved because of their flexibility. (D) A model of talin-1 in direct interaction with Rap1. A linear conformation of talin-1 head domain (green) is displayed in this model. Rap1b is bound to the plasma membrane using its C-terminal geranyl-geranyl moieties. The THD regions (blue) known to interact with the negatively charged PIP2 phospholipids face the membrane: the fly-casting F1 loop; the F2 membrane orientation patch (MOP); and the F3 association patch (FAP). Panels B and C are adapted from Dedden et al33 and panel D from Gingras et al69 with permission.
Domain organization and structure of talin-1. (A) Illustration of autoinhibited (i) and linear (ii) talin-1. Talin-1 consists of a talin head domain (THD) comprising a FERM domain (F0, F1, F2, F3, orange), and a talin rod domain (magenta). (B) Cryo-EM structure of autoinhibited human talin-1. The autoinhibited talin-1 is compactly folded with the talin-1 head interacting with the rod domains. (C) Two intramolecular interactions between the FERM domain and the rod domain, namely F3 (purple)–R9 (mint) and F2 (pink)–R12 (dark green), are shown to be critical for autoinhibition. The F0 and F1 structure is not resolved because of their flexibility. (D) A model of talin-1 in direct interaction with Rap1. A linear conformation of talin-1 head domain (green) is displayed in this model. Rap1b is bound to the plasma membrane using its C-terminal geranyl-geranyl moieties. The THD regions (blue) known to interact with the negatively charged PIP2 phospholipids face the membrane: the fly-casting F1 loop; the F2 membrane orientation patch (MOP); and the F3 association patch (FAP). Panels B and C are adapted from Dedden et al33 and panel D from Gingras et al69 with permission.
Unlike classical FERM domains that fold into a cloverleaf structure,35 crystallographic studies of the talin head alone revealed a linear domain arrangement, which may align parallel to the plasma membrane.36,37 This conformation allows simultaneous binding of the F0 and F1 domains with the membrane-anchored small GTPase Rap1 and interactions of a positively charged loop in the F1 domain and charged residues in the F2 and F3 domains with the membrane phospholipid PIP2 and F3 to the integrin tail (Figure 2D). However, a more recent crystal structure of the talin head in complex with the β3 integrin tail revealed a classical FERM-folded talin head. A C-terminal polylysine motif, which was missing in previous structures, is critically involved in the FERM interdomain contacts required for the cloverleaf structure.38 The FERM domain of talin is more flexible than that of kindlins, but it is unclear whether and how the talin head can extend along the membrane in the active state.
Although talin is considered a master integrin regulator, the fact that integrin activity is completely lost when kindlins are inactivated in cells or animal models indicates that kindlins are crucial regulators of integrin activation (Table 1). The kindlin family consists of 3 members (kindlin-1-3). Kindlin-3 is exclusively found in hematopoietic cells. Kindlin-3–deficent mice die perinatally of anemia, bleeding, and leukocyte adhesion deficiency. Kindlins share high sequence similarity to the talin head; they consist of a FERM domain with an additional F0 domain N terminally added to the F1, F2, and F3 subdomains (Figure 3A).39 Kindlin F3 represents a phosphotyrosine binding–like domain that, unlike talin, binds to the membrane-distal NPxY/F motif of β2 integrin. Kindlins contain a pleckstrin homology (PH) domain inserted into F2. PH domains can target proteins to membranes. Solving the structure of the kindlin-3 PH domain and its lipid-binding properties revealed that the kindlin-3 PH domain binds with higher affinity to phosphatidylinositol (3,4,5)-trisphosphate (PIP3) than PIP2, suggesting that PIP3 patches may cluster kindlin-3 at the membrane.40,41 We recently showed that the PH domain of kindlin-3 is required for its translocation from the cytosol to the plasma membrane and that this translocation precedes β2 integrin activation.42 Consequently, neutrophil arrest in response to chemokine stimulation is completely abolished in cells that express kindlin-3 without the PH domain, similar to kindlin-3–deficient cells.42 Membrane targeting of kindlin-3 is also promoted by a long flexible loop containing a poly-lysine stretch in the F1 domain and a positively charged surface of the F0 domain.43,44 Recent studies showed that the F0 and F2 domains of kindlin-3 also interact with other proteins. The kindlin-3 F0 domain interacts with members of the paxillin family. Paxillin may serve as a bridge between kindlin-3 and talin, which may promote talin head binding to integrin tails. This model may explain how kindlins promote talin-mediated integrin activation.45 Kindlin-3 also directly binds to leupaxin,46 another member of the paxillin family, and recruits it to podosomes, adhesion structures typically found in myeloid cells. Notably, paxillin does not need assistance by kindlin-3 for podosome localization, but kindlin-3 stabilizes podosome lifetime. Thus, in addition to inducing integrin activation through binding to the integrin cytoplasmic tail, kindlin-3 serves as a protein binding platform,46 likely relevant for myeloid cell spreading after arrest.
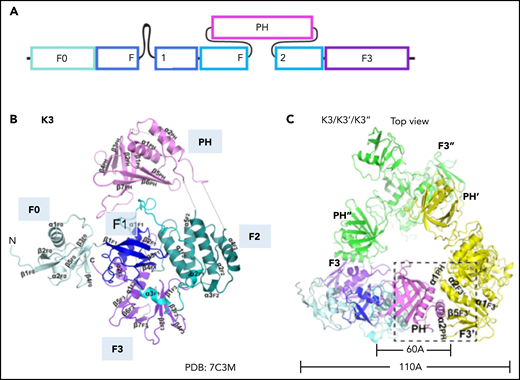
Domain organization of kindlin-3 in the monomer and homotrimer conformation. (A) Domain organization of kindlin-3. Kindlin-3 contains a FERM domain consisting of F0, F1, F2, and F3 subdomains. F1 domain contains a long unstructured loop comprising 83 residues. A pleckstrin homology (PH) domain is inserted in the F2. (B) Crystal structure of kindlin-3 monomer. The kindlin-3 adopts a FERM domain cloverleaf-like conformation. (C) Crystal structure of the kindlin-3 homotrimer. Kindlin-3 PH domain interacts with the F3 subdomain of another kindlin-3 molecule, thereby masking the integrin binding site of kindlin-3. Panels B and C are adapted from Bu et al.51
Domain organization of kindlin-3 in the monomer and homotrimer conformation. (A) Domain organization of kindlin-3. Kindlin-3 contains a FERM domain consisting of F0, F1, F2, and F3 subdomains. F1 domain contains a long unstructured loop comprising 83 residues. A pleckstrin homology (PH) domain is inserted in the F2. (B) Crystal structure of kindlin-3 monomer. The kindlin-3 adopts a FERM domain cloverleaf-like conformation. (C) Crystal structure of the kindlin-3 homotrimer. Kindlin-3 PH domain interacts with the F3 subdomain of another kindlin-3 molecule, thereby masking the integrin binding site of kindlin-3. Panels B and C are adapted from Bu et al.51
Kindlin-3–dependent integrin activation requires posttranslational modifications. Initial mass spectrometric measurements of platelets on agonist stimulation identified phosphorylation of threonine 482 and serine 485 (in mouse kindlin-3), which are critical for platelet integrin activation.47 The same serine at position 484 (in human kindlin-3) was later shown to be phosphorylated by protein kinase C alpha (PKCα)48 in an integrin-linked kinase (ILK)-dependent48 manner. ILK is a pseudo-kinase and a known interacting partner of kindlins.49 PKCα deficiency results in a neutrophil adhesion defect, and PKCα membrane targeting depends on ILK.48 ILK deficiency reduced leukocyte arrest, adhesion, and trans-endothelial migration by half, suggesting that ILK-dependent PKCα-mediated kindlin-3 S485 phosphorylation is involved in integrin activation.48 Because kindlin-3 binds ILK rather weakly,49 it is unclear whether a direct interaction between kindlin-3 and ILK is required for kindlin-3 phosphorylation and integrin activity regulation.
A first structure of recombinantly expressed kindlin-3 studied by X-ray scattering suggested an extended conformation similar to that described for the talin head.50 More recent studies of full-length kindlin-3 and truncated versions of kindlin-2 and 3 lacking the flexible F1 loop and the PH domain displayed the classical cloverleaf conformation of the FERM domain.51-53 A major difference between talins and kindlins appears to be the flexibility of their FERM domains. Although talin’s FERM domains may be able to shift between a cloverleaf and an elongated form, intramolecular interactions between the kindlin F1 and F3 domains keep the FERM domain in a cloverleaf-like structure. Consistent with this, structure-based mutations that disrupt the F1/F3 interface significantly suppressed kindlin-3 activity as measured by platelet integrin activation.53 Recent crystal structures of kindlins also indicated oligomerization of kindlins, which may provide a molecular explanation for the kindlin-induced integrin clustering. A kindlin-2 homodimer was first observed in the crystal structure of a truncated kindlin-2 lacking the PH domain and the F1 loop.52 In this structure, the dimerization was mediated by the F2 domain.52 However, an independent study using a similar truncation strategy revealed that human kindlin-3 without the PH domain and the F1 loop exists only as monomer.53 Nevertheless, forced dimerization of kindlin-3 by fusing kindlin-3 with the dimer-forming GST tag enhanced the talin head domain–induced integrin activation in CHO-A5 cells (expressing platelet αIIbβ3 integrin), which is not observed after monomeric kindlin-3 expression. Whether dimeric kindlin-3 is the active conformation remains to be determined.
Adding kindlin-3 to nanodiscs expressing individual integrin dimers showed no effect,54 but it is unclear whether the recombinant kindlin-3 was functional in these experiments. It is possible that other proteins in hematopoietic cells may promote kindlin-3 dimerization to facilitate integrin activation.53 In addition to the structural studies, a series of biochemical and cell biological analyses revealed that both kindlin-2 and kindlin-3 self-associate into oligomers of 2 to 4 molecules in mammalian cells and that kindlin-3 self-associates to a greater extent than kindlin-2.55 Interaction interfaces for oligomerization were also identified within the F2PH domains.55 However, introduction of equivalent point mutations in the F2 domain of kindlin-3 that perturbed kindlin-2 dimerization had no effect on kindlin-3 self-association. Because the PH domain was lacking in the truncated kindlin-2 dimer, the physiologic significance of the F2-mediated dimerization is questionable. This study also showed that mutations in the F3 domain that impaired self-association enhanced integrin binding, suggesting that kindlin-3 self-association is a negative regulatory mechanism of integrin activation.55 Recently, full-length kindlin-3 was crystalized, revealing a homo-trimer auto-inhibited state.51 In this auto-inhibited conformation, the integrin-binding pocket in the F3 subdomain of each protomer is occluded by the PH domain of another protomer (Figure 3B-C). This means that the kindlin-3 homotrimer must be broken up first, so that the PH domain becomes available for PIP3 binding at the plasma membrane and the F3 domain becomes available for β2 (or β3) integrin cytoplasmic tail binding to induce integrin activation. Mechanisms that trigger kindlin-3 release, such as posttranslational modifications or interaction with other proteins, have yet to be identified. Although this model is very appealing, whether kindlin-3 can trimerize in vivo requires further investigations. However, based on gel filtration, the major population of kindlins in insect and mammalian cell expression systems is monomeric.51,52 Cells such as platelets that express very high kindlin-3 levels might use kindlin-3 self-assembly to prevent spontaneous integrin activation.
New signaling pathways leading to integrin activation
Leukocyte β2 integrins become fully activated on chemokine-triggered inside-out signaling,2,16 in which the small GTPase Rap1 (2 isoforms: Rap1a and Rap1b) plays a central role.56 Active GTP-bound Rap1 binds to several effectors and provides an anchor point for recruiting these proteins to the plasma membrane, as Rap1 is targeted to the membrane by covalent geranyl-geranylation of a cysteine residue.57 The essential role of Rap1 in integrin regulation of hematopoietic cells was demonstrated by Rap1a and Rap1b double knockout mice showing defective leukocyte adhesion58 and abnormal platelet function.56 Both defects are less severe than in the kindlin-3 knockout mice.59 A key function of Rap1 in integrin activation is its role in recruiting cytoplasmic talin-1 to the plasma membrane, which is achieved by 2 different pathways. A ternary integrin activation complex consists of active GTP-bound Rap1, the adapter and Rap1 effector protein RIAM (Rap1 interacting adapter molecule), and talin-1. This complex is particularly important for β2 integrin activation.60-62 Readers are referred to other reviews for a more detailed description of the Rap1-RIAM-talin signaling axis.63 Mechanistically, Rap1-bound RIAM binds talin-1 with its N terminus and recruits it to the membrane by interacting with the talin rod R3 domain and displaces autoinhibitory talin head-rod interactions by binding to the talin-1 F3 domain.64 This process requires that an intermolecular interaction that masks the PIP2-binding site in the PH domain of RIAM is disrupted by Src-dependent phosphorylation, which enables plasma membrane association and integrin activation.65 RIAM-deficient mice display severe defects in β2 integrin-mediated leukocyte adhesion and trafficking. Their platelet functions, however, are normal, because of their very low RIAM expression levels compared with that of talin-1 and Rap1.58,66 RIAM belongs to the MRL (MIG-10, RIAM, and lamellipodin) family of adapter proteins.67 Recently, the RIAM paralogue lamellipodin was shown to compensate for RIAM deficiency in regulatory T cells, whereas conventional T-cell homing to the gut was RIAM dependent.68
The second pathway by which Rap1 recruits talin to the membrane is based on their direct interaction (Figure 2D).69,70 In contrast to the Rap1–RIAM–talin-1 pathway, which is crucial to control leukocyte integrin activation, the direct Rap1–talin-1 pathway represents a general integrin regulatory pathway as it controls talin-1 activity independent of the cell type or integrin class in various organisms such as amoebae, flies, and mice.71-73 Platelet integrin activation completely depends on this pathway, possibly because talin-1, kindlin-3, and Rap1 are very abundant in platelets.74 Initial studies identified a Rap1 binding site within the talin F0 domain.70,71,75 However, platelet integrin activation and function and neutrophil adhesion and extravasation were only slightly affected in talin-1 F0 mutant mice.73 The reason for these mild phenotypes is likely the presence of a second Rap1 binding site in the F1 domain of talin-1.69,73 Notably, although the affinity of talin F0 to active Rap1 is much higher compared with the F1 domain, additional binding of Rap1 to the talin-1 F1 domain is required for efficient talin-1 membrane recruitment in mice but not flies.71,73 Thus, the binding of Rap1 to individual F0 or F1 domains is not sufficient to achieve full talin-1 activation in mammals. Therefore, it is likely that initial contact of the talin-1 F0 domain with active Rap1 recruits talin-1 to the plasma membrane and allows subsequent weaker talin-1 F1–Rap1 interactions. The latter interactions may orient talin-1 in relation to the integrin tail and might be particularly important for efficient integrin activation.69,73 Consistently, conditional talin-1 F0F1 double mutant mice showed a strong defect in talin-1–mediated activation of platelet β1- and β3-integrins.74 Neutrophils carrying the Rap1-binding mutations in the talin-1 F0F1 domains have not been reported yet. Of note, constitutive talin-1 F0F1 double mutant mice are early embryonic lethal, underscoring the importance of the direct Rap1–talin pathway for integrin activation in embryology.69,70
A comparison of the relative roles of the Rap1–talin and the Rap1–RIAM–talin-1 pathways for neutrophil rolling, adhesion, and extravasation was recently conducted in compound mutant mice carrying a RIAM knockout allele and the Rap1 binding site mutation in the talin-1 F0 domain.76 This study revealed a dominant role of the RIAM-dependent pathway on neutrophil rolling and adhesion in vivo. Nevertheless, the Rap1–talin-1 pathway also contributed to leukocyte rolling and adhesion. Although neutrophils from mice with a talin-1 F0 single mutation did not roll faster, double mutant RIAM/talin-1 F0 neutrophils showed greatly increased rolling velocities compared with RIAM mutants. Indeed, the neutrophil rolling velocity of double mutants was comparable to that of talin-1–deficient neutrophils, suggesting that both pathways but no others mediate talin-1 membrane targeting.76 Expression of human β2 integrin in neutrophil-like cells generated from immortalized progenitors isolated from single and double RIAM-deficient and talin-1 F0 mutant mice revealed a synergistic effect on β2 integrin activation as measured by KIM127 and mAb24 binding.76 Thus, it appears that talin-1 in neutrophils is recruited by both pathways. The contribution of the Rap1/talin-1 pathway might be underestimated in this study as Rap1 binding to the talin-1 F1 domain is still possible, and interactions of multiple sites in the talin-head domain bind membrane lipid (PIP2) and stabilize binding of talin to integrins.36,77,78
Whereas the interactions between Rap1 and talin-1, although weak, are apparently sufficient to trigger rapid integrin activation in platelets because of their high expression levels, rapid β2 integrin activation of leukocytes may rely on the synergy of both talin recruitment pathways. However, the ability of Rap1 to regulate β2 integrin activation via 2 distinct effectors, talin-1 and RIAM, also suggests that these 2 signaling pathways have distinct functions in β2 integrin activation in addition to talin recruitment. In particular, β2 integrins depend heavily on the Rap1–RIAM–talin-1 pathway, whereas other integrins do not. Although RIAM recruits talin to the membrane by interacting with the R3 domain of talin-1, interaction with the F3 domain releases autoinhibitory talin head-rod interactions,64 which may be particularly important for efficient β2 integrin activation. In addition, RIAM could provide rapid linkage of β2 integrins to the actin cytoskeleton, either through the subsequent exchange of RIAM with vinculin after talin recruitment or directly via the C-terminal ENA/Vasp binding sites of RIAM.
Mechanisms and functions of talin-1 and kindlin-3 in integrin activation
Resting integrins with a bent-closed conformation have their α and β subunits held together by 2 intersubunit associations (αβ clasps) in the cytoplasmic and transmembrane domains (TMDs). Nuclear magnetic resonance structure analyses of αIIbβ379 and accumulating evidence from structural and functional studies of β1, β2, and β3 integrins80,81 revealed an inner membrane clasp (IMC) including a salt bridge in the membrane-proximal region of the cytoplasmic tails, and an outer membrane clasp in the TMD.79 Disrupting either clasp results in integrin activation.80,82
The prevailing model of integrin activation is that talin-1 binding to the β cytoplasmic domain disrupts the intersubunit associations,83 which alter the position and orientation of the integrin α and β TMD and eventually trigger a change in the extracellular domains from a bent to an extended conformation.84 Integrin tail separation during integrin activation was demonstrated by measuring fluorescence resonance energy transfer between cyan fluorescent protein–fused and yellow fluorescent protein–fused αL and β2 cytoplasmic domains.83 Integrin-activating stimuli led to a decrease in fluorescence resonance energy transfer efficiency. A model supported by nuclear magnetic resonance structure and mutagenesis studies proposes that talin-1 activates integrins by altering the tilt angle of the β TMD, weakening the αβ interaction and resulting in activation of integrins.63,85 This model is supported by the introduction of a flexible kink in the β2 integrin TMD, which impaired chemokine-induced β2 integrin E+ and leukocyte slow rolling and arrest but not the H+ conformation.84 The mechanism underlying this differential effect of introducing a flexible kink is unknown. Notably, the phenotype of neutrophils with the β2 kink mutation is different from kindlin-3 deficiency, which results in partial loss of E+ and complete loss of H+ integrin activation.42 Talin-1 loss causes complete loss of both E+ and H+ conformations of β2 integrins.84 Binding of talin-1 to the membrane proximal NPxY/F motif in the β integrin tail facilitates a second, weaker interaction of a leucine residue (L325) in the talin-1 F3 domain with 2 membrane-proximal Phe residues (F727 and F730) in β3 via hydrophobic interaction.63,86 This interaction seems not to be important for integrin ectodomain E+ but crucial for full activation, as a mutant talin-1 (L325R) was demonstrated to prevent headpiece opening of β2 integrin on neutrophils, with normal slow rolling but defective neutrophil arrest.87 This phenotype is reminiscent of kindlin-3 knockout neutrophils but different from talin-1 knockout neutrophils.88
Thermodynamically, the bent-closed integrin conformation is the most favored conformation. A significant amount of energy is required to shift integrins into the extended closed or open conformations.89,90 Binding of talin (and kindlin) may be sufficient to induce tail separation; however, additional forces may be required to stabilize the extended conformations. Flow-based mechanical forces exerted by the immobilized ligand such as ICAM-1 on endothelial cells are transduced across integrins into the cell and linked to the actomyosin cytoskeleton. During selectin-mediated rolling of leukocytes, PSGL-1 and chemokine-induced signals rapidly trigger talin activation. Unlike kindlin-3, talin-1 has 2 actin binding sites (ABSs) within its C-terminal rod domains R4-R8 and R13-DD91,92 and 1 additional ABS in F2F3 of talin FERM domain.93,94 Thus, activated talin-1 can transmit the force from outside the cell through linkage with the cytoskeleton. The resulting traction forces may help to stabilize the β2 integrin in its extended open conformation (Table 2).
Whether and how kindlin-3 contributes to changes in integrin conformation is less well understood. Deletion of talin-1 affects both neutrophil slow rolling and arrest, whereas neutrophils with kindlin-3 deficiency show normal slow rolling but no arrest. These results suggested that talin-1 is necessary for both the E+ and H+ conformation, whereas kindlin-3 only is required to induce the H+ conformation.88 Indeed, deletion of kindlin-3 completely abolishes the H+ but only partially the E+ integrin conformation.42 In rolling neutrophils, kindlin-3 recruitment to the plasma membrane precedes H+ β2 integrins and cell arrest by almost 20 seconds.42 Kindlin-3 is recruited in broad patches, areas much larger than islands of mAb24-binding H+ integrins. This suggests that kindlin-3 recruitment to the membrane is required but not sufficient to activate β2 integrins. Kindlin-3 likely cooperates with other molecules like talin-1 to activate β2 integrins such that both talin-1 and kindlin-3 are required for full integrin activation. How talin-1 and kindlin-3 cooperate during β2 integrin activation is not clear. As both proteins bind to distinct sites at the β integrin tail independent of each other, simultaneous binding on the same β2 tail may be possible.95 One hypothesis for how kindlin-3 might promote H+ integrin activation82 entails transient kindlin-3 binding to the Asp709-containing membrane-proximal region of the β2 integrin cytoplasmic domain, part of the inner membrane clasp. Thus, kindlin-3 binding may contribute to β2 integrin LFA-1 (αLβ2) activation by unclasping the α/β intersubunit salt bridge82 (Figure 4). However, kindlin-3 seems to be only required to initiate the unclasping. Once unclasped, kindlin-3 becomes dispensable, and talin-1 is sufficient to keep the integrin legs apart. The intersubunit unclasping of the integrins, perhaps initiated by kindlin-3 and maintained by talin-1, followed by mechanical forces through actomyosin-linked talin, may ultimately contribute to the final step of H+ integrin induction. Alternatively, or in addition, kindlin-3 may be involved in integrin clustering, influencing binding avidity but not the single integrin affinity.54
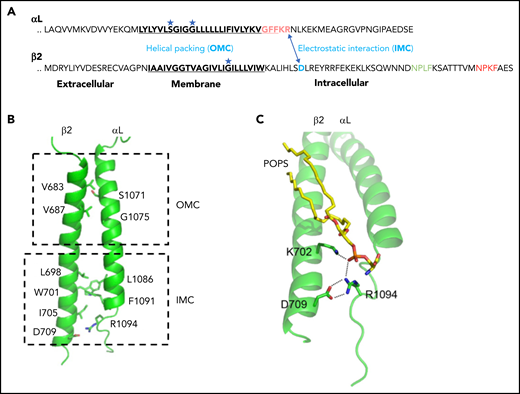
Inner membrane clasp and outer membrane clasp of LFA-1. (A) The inhibitory salt bridge of IMC (electrostatic interaction between the αL-R1094 and β2-D709) and the outer membrane clasp (helical packing) mediated by glycine in the TMDs. (B) Simulated αLβ2 dimer structures. The side chains of major residues involved in dimerization are shown in sticks. (C) Trimeric interaction among αL, β2, and POPS (1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-l-serine). The lipid phosphate group simultaneously engages the β2-K702 amino group and the αL-R1094 guanidino group. Panels B and C are adapted from Guo et al.81
Inner membrane clasp and outer membrane clasp of LFA-1. (A) The inhibitory salt bridge of IMC (electrostatic interaction between the αL-R1094 and β2-D709) and the outer membrane clasp (helical packing) mediated by glycine in the TMDs. (B) Simulated αLβ2 dimer structures. The side chains of major residues involved in dimerization are shown in sticks. (C) Trimeric interaction among αL, β2, and POPS (1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-l-serine). The lipid phosphate group simultaneously engages the β2-K702 amino group and the αL-R1094 guanidino group. Panels B and C are adapted from Guo et al.81
Integrated model of β2 integrin activation
Based on the structural and functional knowledge available today, we propose an integrated model of β2 integrin activation. At rest, PSGL-1 is not engaged, and no chemokine is available. Most or all β2 integrin heterodimers are in the bent, inactive state (Figure 5A). Kindlin-3 is an autoinhibited homotrimer in cytosol, and talin-1 is an autoinhibited ball in the cytosol. Both are away from the plasma membrane (Figure 5A).
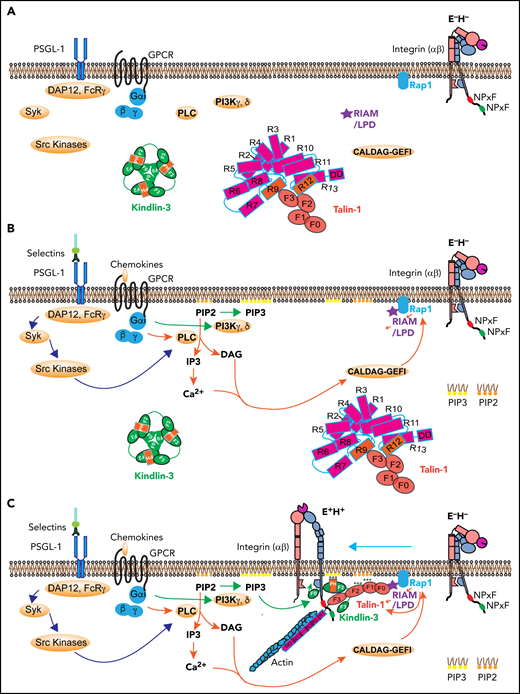
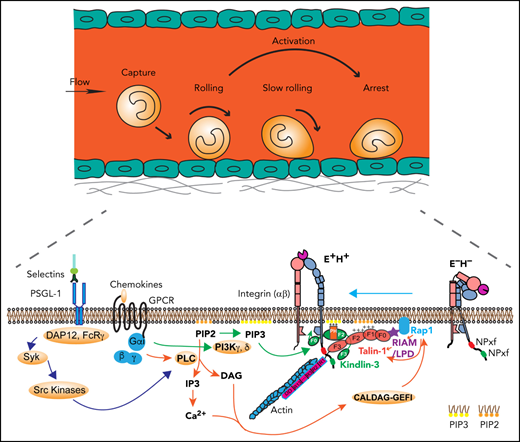
Signaling pathways leading to integrin activation. (A) Integrin at resting state (E−H−), with homotrimeric kindlin-3 and autoinhibited talin-1 in the cytosol. (B) Integrin activation is initiated by 2 signaling pathways: the selectin signaling and chemokine signaling. Engagement of PSGL-1 by selectins triggers serial activation of kinases and recruitment of adaptors. Engagement of chemokine receptors on leukocytes by inflammatory signals leads activation of PLC and PI3K. PLC breaks PIP2 into IP3 and DAG. IP3 further activates IP3 receptors on the endothelium reticulum (ER) and Ca2+ release from the ER store. Rap1 GTPases such as CALDAG-GEFI are activated by Ca2+ and DAG, which stimulates Rap1 bound on the membrane by its C-terminal geranyl-geranyl moieties. PI3K kinases convert PIP2 to PIP3. (C) Rap1 can directly activate talin-1 or indirectly through recruitment of MRL (mig-10/RIAM/lamellipodin) proteins, forming a complex with talin-1. Kindlin-3 is recruited to the plasma membrane using its PH domain. The kindlin-3 PH domain prefers PIP3. Talin-1 and kindlin-3 recruitment to the membrane-proximal compartment and binding to integrin β2 tail are common final steps in triggering integrin activation to adopt the extended high affinity (E+H+) conformations. GPCR, G-protein coupled receptor; LPD, lamellipodin.
Signaling pathways leading to integrin activation. (A) Integrin at resting state (E−H−), with homotrimeric kindlin-3 and autoinhibited talin-1 in the cytosol. (B) Integrin activation is initiated by 2 signaling pathways: the selectin signaling and chemokine signaling. Engagement of PSGL-1 by selectins triggers serial activation of kinases and recruitment of adaptors. Engagement of chemokine receptors on leukocytes by inflammatory signals leads activation of PLC and PI3K. PLC breaks PIP2 into IP3 and DAG. IP3 further activates IP3 receptors on the endothelium reticulum (ER) and Ca2+ release from the ER store. Rap1 GTPases such as CALDAG-GEFI are activated by Ca2+ and DAG, which stimulates Rap1 bound on the membrane by its C-terminal geranyl-geranyl moieties. PI3K kinases convert PIP2 to PIP3. (C) Rap1 can directly activate talin-1 or indirectly through recruitment of MRL (mig-10/RIAM/lamellipodin) proteins, forming a complex with talin-1. Kindlin-3 is recruited to the plasma membrane using its PH domain. The kindlin-3 PH domain prefers PIP3. Talin-1 and kindlin-3 recruitment to the membrane-proximal compartment and binding to integrin β2 tail are common final steps in triggering integrin activation to adopt the extended high affinity (E+H+) conformations. GPCR, G-protein coupled receptor; LPD, lamellipodin.
When endothelial selectins bind PSGL-1, DAP12 and FcRγ are recruited, which leads to activation of Syk and Src kinases (Figure 5B).1,9,10,13,96 Chemokine binding to their G-protein coupled receptor leads to dissociation of Gα from Gβγ, which activates phospholipase C (PLC) and phosphatidylinositol-3-kinase (PI3K) γ and δ (Figure 5B). PLC cleaves PIP2 to inositol 1,4,5-trisphosphate (IP3) and diacylglycerol (DAG). IP3 triggers a calcium (Ca2+) transient, but there is little evidence that the Ca2+ transient is required for integrin inside-out signaling. PI3K γ and δ phosphorylates PIP2 into PIP3, the preferred binding partner of the kindlin-3 PH domain. DAG activates Ca2+ and diacylglycerol-regulated guanine nucleotide exchange factor I (CALDAG-GEFI) , which can activate Rap1.97,98 CALDAG-GEFI deficiency causes reduced β1 integrin– and β2 integrin–mediated adhesion of neutrophils in vitro and has impaired but still residual leukocyte adhesion in vivo,98 suggesting that other GTPases may be also involved in Rap1 activation. Active Rap1, together with PIP2 and RIAM, may recruit and disinhibit talin-1 (Figure 5C). PIP3 may recruit kindlin-3,42 which may transiently break the inner membrane clasp by inserting its F0 domain.82 This unclasping leads to β2 integrin activation if the β2 transmembrane domain is rigid enough.84 Talin-1 apparently can keep the inner clasp open and integrin active. Because talin-1 binds the actin cytoskeleton (Figure 5C), force exerted on the active integrin by the flowing blood is transmitted to the cytoskeleton, which may stabilize the activated integrin conformation. The events shown in Figure 5 are plausible based on the available knowledge. However, not all steps are proven, so this can be considered a working hypothesis.
Conclusions
Over the last decade, we have gained extensive knowledge about the main players of integrin activation and how they interact with each other. Kindlin-3 and talin-1 bind the short cytoplasmic tail of integrins in blood cells. Defining the molecular events that lead to integrin activation will establish molecular mechanisms of integrin activation and provide mechanistic insights into leukocyte recruitment, inflammation, and other immune functions, which may lead to new drug targets that can promote99 or inhibit integrin activation.100
Acknowledgments
The authors thank Alexandre R. Gingras for critical discussions. The authors apologize to those whose studies could not be cited owing to space limitations. Work from the authors’ laboratory was supported by National Institutes of Health grant HL078784 (to K.L.), and L.W. was supported by postdoctoral fellowship 19POST34450228 from the American Heart Association. M.M. was supported by the Deutsche Forschungsgemeinschaft (grant SFB914, TP A01) and Bundesministerium für Bildung und Forschung (grant 01GM1912, eRARE-LADOMICs consortium).
Authorship
Contribution: L.W., M.M., and K.L. wrote and edited the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Lai Wen, La Jolla Institute for Immunology, 9420 Athena Circle Dr, La Jolla, CA 92037; e-mail: lwen@lji.org