Key Points
PUPs A-LONG is the first study of an extended half-life recombinant factor VIII (rFVIIIFc) in PUPs with hemophilia A.
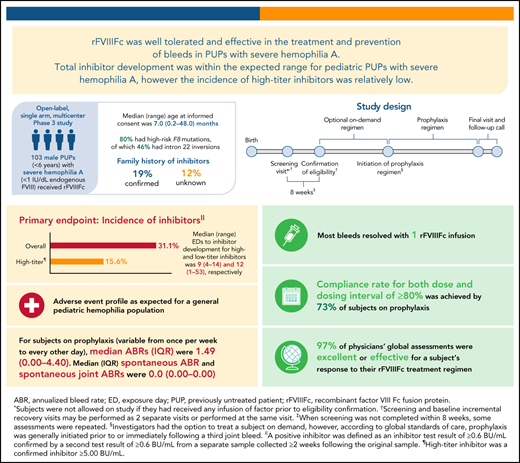
Incidence of inhibitors for patients with ≥10 exposure days was 31.1%; incidence of high-titer inhibitors was relatively low at 15.6%.

Abstract
PUPs A-LONG evaluated the safety and efficacy of recombinant factor VIII Fc fusion protein (rFVIIIFc) in previously untreated patients (PUPs) with hemophilia A. This open-label, phase 3 study enrolled male PUPs (<6 years) with severe hemophilia A to receive rFVIIIFc. The primary endpoint was the occurrence of inhibitor development. Secondary endpoints included annualized bleed rate (ABR). Of 103 subjects receiving ≥1 dose of rFVIIIFc, 80 (78%) were aged <1 year at the study start, 20 (19%) had a family history of inhibitors, and 82 (80%) had high-risk F8 mutations. Twenty subjects began on prophylaxis, while 81 began an on-demand regimen (69 later switched to prophylaxis). Eighty-seven (81%) subjects completed the study. Inhibitor incidence was 31.1% (95% confidence interval [CI], 21.8% to 41.7%) in subjects with ≥10 exposure days (or inhibitor); high-titer inhibitor incidence was 15.6% (95% CI, 8.8% to 24.7%). The median (range) time to high-titer inhibitor development was 9 (4-14) exposure days. Twenty-eight (27%) subjects experienced 32 rFVIIIFc treatment-related adverse events; most were inhibitor development. There was 1 nontreatment-related death due to intracranial hemorrhage (onset before the first rFVIIIFc dose). The overall median (interquartile range [IQR]) ABR was 1.49 (0.00-4.40) for subjects on variable prophylaxis dosing regimens. In this study of rFVIIIFc in pediatric PUPs with severe hemophilia A, overall inhibitor development was within the expected range, although high-titer inhibitor development was on the low end of the range reported in the literature. rFVIIIFc was well-tolerated and effective for prophylaxis and treatment of bleeds. This trial is registered at www.clinicaltrials.gov (NCT02234323).
Introduction
Hemophilia AQ7 A is an X-linked disorder characterized by factor VIII (FVIII) deficiency, with an estimated prevalence of 24.6 cases at birth per 100 000 males, including 9.5 cases for severe hemophilia A.1 Patients with severe hemophilia A (<1 IU/dL [<1%] endogenous FVIII activity) experience spontaneous bleeds and prolonged bleeding following trauma and/or surgery.2 Bleeds, which can be life-threatening, begin in early childhood. Recurrent joint bleeds (hemarthrosis) may lead to long-term, irreversible joint damage with subsequent disability, pain, and significantly reduced quality of life.3-5
The current standard of care (SOC) for patients with hemophilia A is the prevention of bleeds.2 This has been accomplished through prophylaxis with FVIII products (plasma-derived or recombinant).2 Recently, nonfactor prophylaxis has been established, but data in PUPs are missing. Prophylaxis with FVIII concentrate increases circulating FVIII levels, reducing bleed frequency, preserving joint health, and improving quality of life.6,7 To optimize hemostatic control, treatment with standard half-life (SHL) factor products typically requires 3 to 4 IV infusions per week, which is demanding for families and may negatively impact adherence to treatment owing to the burden.8 In children, frequency of factor administration may necessitate the use of central venous access devices (CVADs).9 Extended half-life (EHL) FVIII concentrates were developed to reduce the treatment burden by allowing for less frequent infusions and/or to enable more optimal protection by maintaining higher factor levels.10
Inhibitor development, which occurs in 25% to 40% of previously untreated patients (PUPs) with severe hemophilia A exposed to SHL products,11-16 is the most serious complication of hemophilia treatment because it renders FVIII replacement ineffective for treatment or prevention of bleeds. Patients with hemophilia tend to develop inhibitors within 20 exposure days (EDs), although some occur up to the 75th ED.17 Established genetic risk factors for inhibitors include null F8 gene mutations and family history of inhibitors.18,19
The availability of a nonfactor product has provided an alternative prophylactic option for patients with hemophilia A and inhibitors, but FVIII replacement is still required for breakthrough bleeds and management of surgical hemostasis. Consequently, developing an inhibitor continues to be an undesired complication of FVIII exposure in young children. To date, rates of inhibitor development among PUPs receiving EHL FVIII therapies have not been reported. This is important as these products may be associated with more, less, or the same inhibitor development as seen with SHL FVIIIs.
Recombinant FVIII Fc fusion protein (rFVIIIFc) (Eloctate and Elocta, Sanofi, Waltham, MA, and Sobi, Stockholm, Sweden) was the first EHL FVIII therapy approved for prophylaxis as well as on-demand (OD) treatment of bleeding episodes and perioperative bleed management in the United States, Europe, and other countries in patients of all ages with hemophilia A. Manufactured in a human cell line, rFVIIIFc consists of a single molecule of recombinant FVIII (rFVIII) fused to the Fc domain of human immunoglobulin G1, enabling recycling through the neonatal Fc receptor, thereby prolonging its half-life.20-22
Clinical trials with rFVIIIFc have demonstrated low annualized bleed rates (ABRs) in previously treated patients (PTPs) with severe hemophilia A ≥12 years of age treated every 3 to 5 days or once weekly (A-LONG [NCT01181128])23; rFVIIIFc was also safe and efficacious in a pediatric PTP population (<12 years of age) with severe hemophilia A (Kids A-LONG [NCT01458106]).24 Long-term safety and efficacy assessments (up to 5.9 years cumulative duration) from the extension study confirm rFVIIIFc to be safe and effective at providing low ABR with extended dosing intervals in PTPs.25 The PUPs A-LONG study (NCT02234323) aimed to investigate rFVIIIFc safety (main objective: establishing inhibitor risk) and efficacy in pediatric PUPs with severe hemophilia A. Herein, we report the results of this study.
Methods
Study design
PUPs A-LONG (NCT02234323) was an open-label, multicenter, phase 3 study that enrolled male pediatric PUPs with severe hemophilia A to receive rFVIIIFc (supplemental Figure 1, available on the Blood Web site). The study was performed in accordance with the Declaration of Helsinki and all local regulations. Written informed consent was obtained from the subjects’ parents or legal guardians.
Investigators had the option to treat subjects episodically until a prophylactic regimen was initiated in accordance with the local SOC. Given global SOCs, it was recommended that prophylaxis be initiated before or, at the latest, immediately following a third episode of hemarthrosis. The recommended initial dose for prophylaxis was 25-80 IU/kg at 3- to 5-day intervals, with adjustments based on pharmacokinetic data, physical activity, and bleed patterns. Samples for FVIII activity were taken pre and postdose every 12 weeks for subjects on prophylaxis. The treatment period was ≥50 EDs to rFVIIIFc, unless the end of the study was declared or study withdrawal occurred. One ED was defined as a 24-hour period in which a participant received ≥1 rFVIIIFc dose, with the time of the first infusion marking the start of the ED.
Subjects were considered to have completed treatment if they reached ≥50 EDs of rFVIIIFc without inhibitor development, met early withdrawal criteria, or the end of the study was reached. Subjects who developed a low-titer inhibitor were eligible to continue at the same or higher dose per infusion of rFVIIIFc at the investigator’s discretion. Those who developed a high-titer inhibitor or low-titer inhibitor with poorly controlled bleeds (assessed by the investigator) despite increased rFVIIIFc dose or if bypassing agents were required to treat bleeds were eligible to receive immune tolerance induction (ITI) with rFVIIIFc. The current analysis focuses on subjects who received either OD or prophylactic treatment with rFVIIIFc.
Subject eligibility
Males <6 years of age at informed consent with severe hemophilia A, defined as <1 IU/dL (<1%) endogenous FVIII, with no prior exposure to blood components or infusion with an FVIII concentrate (including plasma-derived and rFVIII), were enrolled. The main exclusion criteria were history of positive inhibitor testing, infusion with commercially available rFVIIIFc at any time before the screening, other coagulation disorder(s), and any concurrent clinically significant major disease.
Outcomes
The primary endpoint was the occurrence of inhibitor development. A positive inhibitor was defined as an inhibitor test result of ≥0.6 Bethesda units (BU)/mL confirmed by a second test result of ≥0.6 BU/mL from a separate sample, drawn 2 to 4 weeks following the original sample. Both tests must have been performed by the central laboratory using the Nijmegen-modified Bethesda assay. Secondary endpoints included ABR (overall, spontaneous, traumatic, and spontaneous joint), the total number of EDs, total annualized rFVIIIFc consumption, number of infusions and rFVIIIFc dose per infusion required to resolve a bleeding episode, assessment of rFVIIIFc treatment response by a caregiver for any bleeding event by use of a 4-point bleeding response scale (excellent, good, moderate, and none), and physician assessment of subject’s response to rFVIIIFc treatment regimen by use of a 4-point scale (excellent, effective, partially effective, and ineffective), both of which were assessed every 12 weeks during the study. rFVIIIFc incremental recovery was an additional secondary endpoint, with peak and trough samples collected at baseline and during scheduled clinic visits if the subject received a rFVIIIFc infusion during the visit and was not experiencing a bleed.
Statistical analysis
Demographics and baseline characteristics were summarized using descriptive statistics. Genotype classification was performed according to Gouw and colleagues,16 with high risk encompassing inversions, large deletions (>50 base pairs), and frameshift and nonsense mutations, and low risk encompassing splice site and missense mutations. There were 2 exceptions based on the association with moderate or severe hemophilia and inhibitor development: a splice site variant (HGVS [Human Genome Variation Society] F8 c.5999-1G>T) in intron 18 of the FVIII gene and a missense variant (HGVS F8 c.6683G>A) in exon 24 of the FVIII gene, each being classified as high risk (http://www.factorviii-db.org). Safety analyses were based on the safety analysis set, which consisted of all patients who received ≥1 dose of rFVIIIFc. The full analysis set (FAS) was defined as all enrolled patients who received ≥1 dose of study rFVIIIFc; patients were considered enrolled when the investigator confirmed eligibility according to inclusion criteria. Efficacy analyses were based on the FAS.
The primary analysis of the incidence of inhibitor formation was based on all subjects who reached ≥10 EDs, had inhibitor tests performed at or beyond this milestone, or had inhibitor development. Inhibitor testing was conducted at ED milestones (5, 10, 20, 50, 75, and 100 EDs), every 12 weeks (regardless of exposure), or at suspected inhibitor development. Inhibitor incidence was determined based on the inclusion of all subjects who developed an inhibitor after the initial rFVIIIFc administration, regardless of the number of EDs. Incidence of inhibitor formation based on all treated subjects and subjects who had reached ≥20 or ≥50 EDs and had ≥1 inhibitor test performed at or beyond this milestone were also assessed. The cumulative incidence of inhibitors over time was estimated using the Kaplan-Meier method.
For a patient to have an evaluable efficacy period over the duration of the study, he must have ≥1 day of treatment for an OD regimen or ≥2 prophylactic infusions for a prophylaxis regimen in the FAS. Evaluation of efficacy endpoints was performed in the efficacy period. For exposure assessments, the total number of EDs with rFVIIIFc per participant were categorized as <5, 5 to <10, 10 to <20, 20 to <50, 50 to <75, 75 to <100, and ≥100. The total number of infusions per subject was summarized by treatment regimen. Duration of rFVIIIFc dosing and ABR were summarized using descriptive statistics. A sensitivity analysis of ABR was conducted on subjects with ≥50 EDs on a prophylactic regimen, given that some patients were observed for a shorter time. Caregiver and physician assessment of response to treatment were summarized by descriptive statistics.
Treatment adherence was assessed during the efficacy period and summarized for treatment of bleeding episodes and prophylaxis. For individual bleeding episodes, patients were considered compliant if they received rFVIIIFc within 8 hours of bleed onset. For prophylaxis, a dose was considered compliant if it was within 80% to 125% of the prescribed dose, and the dosing interval was considered compliant if the time between 2 prophylactic doses was within ±24 hours of the prescribed dosing interval. Subjects were considered dose compliant or dosing interval compliant if respective rates were ≥80%.
Results
Of 108 subjects enrolled in the study, 103 received ≥1 dose of rFVIIIFc, and 101 had an efficacy period (supplemental Figure 2). Thus, 103 subjects constituted the safety analysis set, and all of these 103 subjects were deemed to have met all study enrollment criteria and thus constituted the FAS.
Eighty-one subjects started the study OD and 20 subjects on prophylaxis. Sixty-nine subjects receiving OD rFVIIIFc switched to prophylaxis, resulting in a total of 89 subjects being on prophylaxis during the study. Eighty-seven (81%) of 108 enrolled subjects completed the study, while 21 (19%) subjects terminated the study early; reasons included physician decision (n = 5), central laboratory results (FVIII ≥1 IU/dL [≥1%]) (n = 3), lack of efficacy (n = 3 [during ITI]), withdrawal of consent (n = 2), death (n = 1), and other (n = 7; unavailable homecare [n = 1], patient identified as meeting an exclusion criterion [n = 2], discontinuation for need of continuous infusion [n=3: 2 for intracranial hemorrhage and 1 for planned surgery], parent decision [n = 1]).
Subject baseline characteristics
Baseline characteristics are shown in Table 1. Most subjects (n = 80 [78%]) were <1 year of age at study enrollment. The median (range) age was 7.0 (0.2-48.0) months at the time of enrollment, with 42 (41%) being <6 months of age.
More than half of the participants (n = 58 [56%]) had received vaccines, antibiotics, antipyretics, or other medications that were considered typical for this population. Of 103 subjects, 20 (19%) had a family history of inhibitor development, and 71 (69%) had no family history of inhibitors, while for 12 subjects (12%), this was unknown. Eighty-two (80%) subjects had predicted high-risk mutations, including 47 subjects (46%) who had intron 22 inversions.16
rFVIIIFc exposure
For PUPs A-LONG, the overall median (range) number of weeks on the study was 64.2 (0.0-206.8), including those individuals who received ITI. The median (range) duration of OD and prophylaxis treatment regimens was 23.6 (0.4-107.8) and 44.0 (0.0-96.6) weeks, respectively. A summary of EDs to rFVIIIFc by treatment regimen is in Table 2. The median (range) of EDs was 4 (0-27) and 91 (1-192) for subjects while OD and while on prophylaxis, respectively.
Minor surgeries
Forty-two of 103 subjects (41%) had CVADs during the study. Eighteen (64%) subjects in the inhibitor subgroup (n = 28) and 24 (32%) subjects in the noninhibitor subgroup (n = 75) had CVADs. Most CVADs were placed in subjects <1 year of age. Four subjects underwent circumcision during the study. Other surgeries included arthrocentesis, tympanostomy, operative dentistry, and penoplasty in 1 subject each.
Inhibitor development
The overall incidence of inhibitor development was 31.1% (n = 28; 95% confidence interval [CI], 21.8% to 41.7%) for participants with ≥10 EDs or who had an inhibitor (Table 3). The incidence of high-titer inhibitors was the same as the incidence of low-titer inhibitors at 15.6% (n = 14; 95% CI, 8.8% to 24.7%). For subjects reaching ≥20 and ≥50 EDs, inhibitor development occurred in 31.5% (95% CI, 22.0% to 42.2%) and 32.6% (95% CI, 22.8% to 43.5%) of subjects, respectively.
The overall median (range) EDs to inhibitor development was 9 (1-53). Median (range) EDs to inhibitor development for high- and low-titer inhibitors was 9 (4-14) and 12 (1-53), respectively. One participant developed an inhibitor after 50 EDs; this subject developed a low titer inhibitor at 53 EDs. The cumulative incidence of inhibitor development estimated using the Kaplan-Meier method was 17.4% at 10 EDs, 26.3% at 20 EDs, and 29.8% at 50 EDs (Figure 1). The cumulative incidence of high-titer inhibitor development was 11.3% at 10 EDs and 16.1% at both 20 and 50 EDs.

Incidence of inhibitor development by EDs and titer. The cumulative incidence of inhibitor development was estimated using the Kaplan-Maier method among those in the safety analysis set (n = 103) according to titer; a high-titer inhibitor was a confirmed inhibitor ≥5 BU/mL, and a low-titer inhibitor was a confirmed inhibitor ≥0.6 and <5 BU/mL. For patients without an inhibitor, follow-up time was censored at the last ED at the time of analysis. The table contains the estimated cumulative incidence of inhibitor development at the 10, 20, and 50 ED milestones.
Incidence of inhibitor development by EDs and titer. The cumulative incidence of inhibitor development was estimated using the Kaplan-Maier method among those in the safety analysis set (n = 103) according to titer; a high-titer inhibitor was a confirmed inhibitor ≥5 BU/mL, and a low-titer inhibitor was a confirmed inhibitor ≥0.6 and <5 BU/mL. For patients without an inhibitor, follow-up time was censored at the last ED at the time of analysis. The table contains the estimated cumulative incidence of inhibitor development at the 10, 20, and 50 ED milestones.
At the time of inhibitor development, 7 subjects were receiving rFVIIIFc OD, and 21 subjects were receiving prophylaxis.
Nine of 20 (45%) subjects with a family history of inhibitors developed inhibitors, compared with 15 of 71 (21%) subjects without a family history of inhibitors; 4 subjects had an unknown family history (Table 4). Of the 9 subjects who developed an inhibitor and had a family history of inhibitors, 6 (67%) developed high-titer inhibitors (supplemental Table 1). Of 82 subjects with an adjudicated high-risk genotype,16 23 (28%) developed inhibitors, and of 5 subjects with an adjudicated low-risk genotype, 1 (20%) developed an inhibitor. Among the 28 subjects that developed inhibitors, 14 had an intron 22 inversion (8 developed a low- and 6 a high-titer inhibitor). Five of 14 (36%) Black or White Hispanic subjects developed inhibitors, compared with 23 of 89 (26%) non-Black or Hispanic subjects.
No subjects underwent major surgery; therefore, associated inhibitor development risk was not assessed.
Of the 28 subjects with inhibitors, 15 received ITI with rFVIIIFc (including 12 with high-titer inhibitors and 3 with low-titer inhibitors), and 2 with high-titer inhibitors were withdrawn from the study (1 due to physician decision and 1 due to lack of home health care). The 11 remaining subjects all had low-titer inhibitors; 7 completed the study (achieved 50 EDs) and achieved remission (defined as 2 consecutive negative inhibitor titers ≥28 days apart) without ITI, 2 completed the study without inhibitor remission during the study, and 2 discontinued (1 had a treatment-emergent adverse event [TEAE] of ICH and 1 was subsequently identified as being ineligible owing to exposure to blood components or FVIII replacement products any time before or during screening).
Adverse events
Of 81 subjects who received rFVIIIFc OD at any point, 71 (88%) experienced ≥1 TEAE, while 75 (84%) subjects receiving rFVIIIFc as prophylaxis experienced ≥1 TEAE (Table 5). TEAEs of infection occurred in 40 (49%) and 64 (72%) of subjects in OD and prophylactic regimens, respectively, with nasopharyngitis most common (n = 11 [14%]) in the OD regimen and upper respiratory tract infection most common (n = 21 [24%]) on prophylaxis.
Sixty treatment-emergent serious adverse events (TESAE) occurred among 38 (47%) subjects while OD; 71 TESAEs occurred in 34 (38%) subjects on prophylaxis. Seven (9%) subjects while OD developed inhibitors, while 21 (24%) subjects receiving rFVIIIFc as prophylaxis experienced FVIII inhibitor development. Other related SAEs included 1 event of deep vein thrombosis (related to an indwelling central venous catheter in a high-titer inhibitor subject on ITI who received bypassing agent after a fall), 1 event of soft tissue hemorrhage (occurred in the context of high-titer inhibitor development), and 1 event of a CVAD-related thrombosis. The CVAD-related thrombosis occurred in a subject who experienced staphylococcal bacteremia and port infection in the days leading up to the event. The thrombotic event related to the indwelling catheter resolved after port removal, and the subject remained in the study.
Six (6%) subjects discontinued the study owing to an adverse event (2 subjects for FVIII inhibitor development, 3 subjects because of an ICH, and 1 subject for poor venous access), all of which were serious. One subject died of ICH, unrelated to the study drug, as he had not received any study drug before the event. The other 2 events of ICH resolved with treatment and were considered unrelated to the study drug; both occurred in subjects receiving OD treatment. There were no reports of anaphylaxis. There was 1 report of hypersensitivity/allergy associated with rFVIIIFc: the subject developed a papular rash, considered nonserious and related to study treatment by the investigator, and resolved without treatment.
rFVIIIFc consumption and prophylactic regimens over time
The median (interquartile range [IQR]) annualized rFVIIIFc consumption was 197.6 (24.4-698.3) IU/kg for subjects while OD and 5384.4 (3627.0-7246.5) IU/kg for subjects while on prophylaxis. Over the time on prophylaxis, the median (IQR) average weekly dose for subjects (n = 88) was 101.4 (63.0-138.7) IU/kg, with a median (IQR) average dosing interval of 3.9 (3.3-4.7) days.
Of the 89 subjects that were on prophylaxis (20 at the start of the study and 69 later switched from OD), most (n = 51 [57%]) started prophylaxis with once per week dosing, receiving a median (range) of 40.0 (20.0-100.0) IU/kg. Yet, for those patients who reached ≥50 EDs during the study, a large proportion (n = 35 [44%]) were receiving twice-weekly dosing with a median (range) dose of 50 (25.0-95.0) IU/kg at or before 50 EDs.
rFVIIIFc incremental recovery
The median (IQR) incremental recovery for the baseline pharmacokinetic profile of those OD was 2.2 (1.9-2.9) IU/dL per IU/kg. For those on prophylaxis, the incremental recovery was 2.1 (1.7-2.7) IU/dL per IU/kg. Incremental recovery was consistent across the duration of the study.
ABR
Table 6 shows ABR by treatment regimen. The median (IQR) overall ABR (while on any prophylaxis regimen) was 1.49 (0.00-4.40), spontaneous ABR was 0.00 (0.00-0.00), traumatic ABR was 0.83 (0.00-3.43), and spontaneous joint ABR was 0.00 (0.00-0.00). The most common site of bleeds in those on prophylaxis was the skin or mucosa (n = 42 [47%]); 23 (26%) subjects had joint bleeds.
Treatment compliance (adherence)
For those on prophylaxis, a compliance rate for both dose and dosing interval of ≥80% was achieved by 65 (73%) subjects, whereas a dose compliance rate ≥80% was achieved by 73 (82%) subjects and a dosing interval compliance rate ≥80% was achieved by 79 (90%) subjects.
Response to rFVIIIFc treatment: number of infusions and dose required to resolve a bleeding episode
The median number of infusions required for bleed resolution for each treatment regimen was 1.0, with an IQR of 1.0 to 2.0 and 1.0 to 1.0 for OD and prophylactic regimens, respectively. Of 148 patients, 107 (72%) and 172/215 (80%) bleeds in the OD and prophylaxis groups, respectively, were resolved with 1 rFVIIIFc infusion; 132/148 (89%) and 201/215 (94%) bleeding episodes, respectively, required ≤2 infusions for resolution in the 2 groups. In the OD group, 16 (11%) bleeds required ≥3 infusions for resolution, compared with 14 (7%) bleeds in the prophylaxis group.
With regards to the dose of rFVIIIFc required to resolve bleeding episodes, the median (IQR) total dose per bleeding episode was 54.5 (34.5-78.4) IU/kg in the OD group and 55.6 (43.1-75.0) IU/kg in the prophylactic group. Per subject, the median (IQR) average dose per infusion was 41.1 (32.6-51.6) IU/kg and 49.0 (41.7-61.5) IU/kg for those on the OD and prophylactic regimens, respectively.
For infusions with a caregiver’s assessment, the subject’s response to infusions, as assessed using a 4-point bleeding response scale, was considered excellent or good for 102/120 (85%) OD infusions and 163/204 (80%) prophylaxis infusions.
Assessment of response to rFVIIIFc treatment regimen
Most physician assessments (97%) of a subject’s response to the rFVIIIFc treatment regimen were excellent or effective.
Discussion
This was the first prospective assessment of an EHL rFVIII in a globally representative population of PUPs with severe hemophilia A, with most being <1 year old at enrollment, with a medical and surgical history typical of a pediatric hemophilia A population in the developed world. The study enrolled a relatively high proportion of subjects with a family history of inhibitors (19%).26,27 Subjects were treated according to local SOC, some beginning an OD regimen and transitioning to prophylaxis as deemed necessary by their hematologist, while others commenced prophylaxis at study entry.
The incidence of inhibitor development was 31.1% (28 of 90 subjects) in subjects with ≥10 EDs, with high- and low-titer inhibitors each developing in 15.6% of subjects. The inhibitor incidence is within the expected range for PUPs with severe hemophilia A (15% to 50%).11-16 The cumulative high-titer inhibitor incidence with rFVIIIFc at 50 EDs (16.1%) was low in relation to previous reports for other FVIII products and other classes of products,12,13,15,16,28,29 for example, the rFVIII arm in the SIPPET (Survey of Inhibitors in Plasma-Product Exposed Toddlers) study (28.4% [95% CI, 19.6% to 37.2%]),13 and among FVIII products in the PedNet (Pediatric Network on hemophilia management) study (22.4%).16 The incidence of high-titer inhibitor development in this study with rFVIIIFc was similar to that of plasma-derived FVIII replacement products in SIPPET (18.6% [95% CI, 11.2% to 26.0%]).13 However, the direct interstudy comparison may not be feasible; study populations between PUPs A-LONG and SIPPET are different geographically and ethnically and racially. Seven patients in the low-titer inhibitor group remained in the study and experienced inhibitor remission without ITI; a similar observation was made in the Real-life Management of Inhibitors (REMAIN) study, where patients with low-titer inhibitors experienced resolution of inhibitors without ITI.30
Preclinical evidence suggests that the Fc domain of rFVIIIFc exerts immunomodulatory effects, which may contribute to the lower rate of high-titer inhibitor development observed in this study; however, this has not been proven clinically.31-33
The median time to inhibitor development was 9 EDs, which aligns with data indicating patients are at the highest risk for inhibitor development in the first 20 EDs, but the residual risk can persist to 75 EDs.16,17,28,29 The development of high-titer inhibitors occurred up to 14 EDs, with no reports of high-titer inhibitors after 14 EDs. In contrast, low-titer inhibitor development occurred up to 53 EDs.
Evidence presented here suggests family history and genotype are risk factors associated with inhibitor development18; however, other descriptive risk factors such as race or ethnicity did not have a discernible association with inhibitor development. One limitation of this study that may impact this result is that it included few Black or Hispanic participants and may not have detected a trend. Vaccination history, infection, or surgery before inhibitor development also did not impact inhibitor incidence in this population. In a report from the Centers for Disease Control and Prevention Universal Data Collection System in children ≤2 years of age with hemophilia, inhibitors were strongly associated with ICH, CVAD use, and CVAD complications.9 Such trends were not observed in the present study; while a higher rate of CVAD insertion was observed in the inhibitor subgroup, many were following inhibitor development. This could be related to the EHL status of rFVIIIFc permitting less frequent infusions and thereby making CVAD insertion less necessary, although further study would be required to confirm this.
rFVIIIFc was well-tolerated in this PUP population without unanticipated safety findings. The type and incidence of adverse events in the study were similar to those expected for the general hemophilia population, particularly the pediatric hemophilia population.
rFVIIIFc was effective for the treatment of bleeds as well as for prophylaxis. Low ABRs were achieved in a young population with high FVIII clearance with prophylactic regimens of low-frequency dosing (weekly) or intermediate to high-frequency dosing (≥2 infusions per week), with the average being 2 infusions per week by 50 EDs. Similar to results in PTPs,23,24 most bleeding episodes required ≤2 infusions to resolve, and the median number of infusions required for resolution was 1.0.
This first prospective study of an EHL rFVIII treatment for PUPs with severe hemophilia A demonstrates overall inhibitor development within the expected range, albeit with a high-titer inhibitor incidence lower than that reported in other studies.11-16 Incremental recovery values were consistent with PTPs on rFVIIIFc and with other FVIII products. rFVIIIFc was well-tolerated and effective for the control and prevention of bleeds in this pediatric PUPs population.
Acknowledgments
The authors, Sanofi, and Sobi gratefully thank the families and all investigators for participating in the PUPs A-LONG study. The PUPs A-LONG study was sponsored by Sanofi (Cambridge, MA) and Sobi (Stockholm, Sweden). Programming support was provided by Peter Loonan, Sanofi Programming Lead, and by Vita Data Sciences. Ekta Seth Chhabra (Sanofi), Suresh Katragadda (Sanofi), and Samuel Lessard (Sanofi) contributed to the data interpretation. The authors also thank Graham Neill (Sanofi) for support and providing critical feedback during manuscript development, as well as Raina Liesner for her input during manuscript development. Medical writing was provided by Ashleigh Pulkoski-Gross and Jennifer Alexander, Fishawack Communications Ltd, part of Fishawack Health (Conshohocken, PA), and this support was funded by Sanofi and Sobi.
Authorship
Contribution: C.K., M.C.O., A.D., R.K., B.N., S.A.B., M.S., and M.C. collected and interpreted data and provided critical revision of the manuscript; S.G., S.M., D.J., and B.W. analyzed and interpreted data and provided critical revision of the manuscript; and all authors had final approval of the manuscript for publication.
Conflict-of-interest disclosure: C.K. has received personal fees from Bayer Vital GmbH, CSL Behring, Novo Nordisk, Roche/Chugai, Sanofi/Sobi, and Takeda; C.K.’s institution has also received grants from Bayer Vital GmbH, Biotest, CSL Behring, Intersero, Novo Nordisk, Pfizer, Sanofi/Sobi, and Takeda. M.C.O. has received research support from Bioverativ/Sanofi, BioMarin, Novo Nordisk, Pfizer, Roche, and Shire/Takeda; received honoraria for speaking/participating in advisory boards from Bioverativ/Sanofi, BioMarin, Pfizer, Roche, and Shire/Takeda. A.D. has received research support from Biogen/Sanofi, BioMarin, and Takeda; received personal fees from Genentech, Medscape, and UniQure and nonfinancial support from Hemophilia Federation of America and International Prophylaxis Study Group. R.K. has served on advisory boards for Bioverativ/Sanofi, BPL, Genentech, Kedrion, Novo Nordisk, Octapharma, Pfizer, Shire/Takeda, and Catalyst Bioscience; has also received clinical trial research grants from Sanofi/Bioverativ, Bayer, BioMarin, Shire/Takeda, Novo Nordisk, and UniQure. B.N. has received personal fees from Sobi and sponsorship from Bayer, CSL Behring, and Sanofi. M.S. has received personal/speaker fees from Bayer, CSL Behring, Kedrion, Roche/Chugai, and Sobi; has also served on advisory boards for CSL Behring, Novo Nordisk, Roche/Chugai, and Sobi. S.G., S.M., and D.J. are employees of Sanofi and may hold shares and/or stock options in the company. B.W. is an employee of Sobi. M.C. reports having received research support from Bayer, Bioverativ/Sanofi, CSL Behring, Novo Nordisk, Octapharma, Pfizer, and Shire/Takeda; has also received honoraria for speaking/participating in advisory boards from Bayer, Bioverativ/Sanofi, Biotest, CSL Behring, Grifols, LFB, Novo Nordisk, Octapharma, Pfizer, Roche, and Shire/Takeda. The remaining authors declare no competing financial interests.
Correspondence: Christoph Königs, University Hospital Frankfurt, Theodor Stern Kai 7, 60596 Frankfurt am Main, Germany; e-mail: christoph.koenigs@kgu.de.
Qualified researchers may request access to patient-level data and related documents (eg, the clinical study report, study protocol with any amendments, blank case report form, statistical analysis plan, and dataset specifications). Patient-level data will be anonymized, and study documents will be redacted to protect the privacy of trial participants. Further details on Sanofi’s data-sharing criteria, eligible studies, and process for requesting access can be found at https://vivli.org/.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.