Key Points
Heterozygous variants in CLPB, clustered around the ATP-binding pocket, are a newly described and common cause of SCN.
ATP-binding pocket CLPB mutants act in a dominant-negative manner to impair mitochondrial function and disrupt granulocytic differentiation.

Abstract
Severe congenital neutropenia is an inborn disorder of granulopoiesis. Approximately one third of cases do not have a known genetic cause. Exome sequencing of 104 persons with congenital neutropenia identified heterozygous missense variants of CLPB (caseinolytic peptidase B) in 5 severe congenital neutropenia cases, with 5 more cases identified through additional sequencing efforts or clinical sequencing. CLPB encodes an adenosine triphosphatase that is implicated in protein folding and mitochondrial function. Prior studies showed that biallelic mutations of CLPB are associated with a syndrome of 3-methylglutaconic aciduria, cataracts, neurologic disease, and variable neutropenia. However, 3-methylglutaconic aciduria was not observed and, other than neutropenia, these clinical features were uncommon in our series. Moreover, the CLPB variants are distinct, consisting of heterozygous variants that cluster near the adenosine triphosphate-binding pocket. Both genetic loss of CLPB and expression of CLPB variants result in impaired granulocytic differentiation of human hematopoietic progenitor cells and increased apoptosis. These CLPB variants associate with wild-type CLPB and inhibit its adenosine triphosphatase and disaggregase activity in a dominant-negative fashion. Finally, expression of CLPB variants is associated with impaired mitochondrial function but does not render cells more sensitive to endoplasmic reticulum stress. Together, these data show that heterozygous CLPB variants are a new and relatively common cause of congenital neutropenia and should be considered in the evaluation of patients with congenital neutropenia.
Introduction
Severe congenital neutropenia (SCN) is a rare bone marrow failure syndrome that is characterized by severe chronic neutropenia, an arrest of granulocytic differentiation at the promyelocyte or myelocyte stage, and a marked propensity to develop myeloid malignancies.1 It has an estimated prevalence of 5 cases per 1 million individuals. SCN demonstrates multiple modes of inheritance, including autosomal-recessive, autosomal-dominant, X-linked, and sporadic patterns. The most frequently mutated gene in SCN is ELANE, which accounts for ∼60% of SCN cases. ELANE mutations are also found in the majority of cases of cyclic neutropenia, a related disorder of granulopoiesis that is characterized by recurrent episodes of neutropenia with a 14- to 35-day periodicity. The genetic cause of ∼30% of cases of SCN remains unknown.
Several groups recently reported that biallelic mutations of CLPB are associated with a syndrome characterized by 3-methylglutaconic aciduria (3-MGA), cataracts, neurologic disease, and neutropenia.2-5 Neutrophils counts are variable, ranging from normal to chronic severe neutropenia. The CLPB mutations are distributed across the entire gene and include frequent non-sense or frameshift mutations,6 suggesting a loss-of-function mechanism of disease pathogenesis. CLPB encodes for caseinolytic peptidase B homolog, a member of the Clp/heat shock protein-100 family of adenosine triphosphatases (ATPases).7 Prokaryotic ClpB has been shown to catalyze protein unfolding and disaggregation in the setting of cellular stress.8-10 This function is dependent on adenosine triphosphate (ATP) hydrolysis and the formation of a hexameric ring through which substrate proteins are driven. Although the ATPase and disaggregase function of human CLPB has been confirmed,11,12 and it does appear to form higher-order multimers,12 the exact structural basis for its function may differ because vertebrate CLPB only shares homology with the prokaryotic C-terminal ATPase domain and has a unique N-terminal region.2 CLPB localizes to mitochondria,2,11,13,14 and it has been shown to regulate mitochondrial function.14
Here, we report exome sequencing results of a large cohort of persons with congenital neutropenia. We identified heterozygous variants in CLPB that cluster in the ATP-binding pocket in 10 unrelated individuals. Expression of CLPB variants results in impaired granulocytic differentiation of human hematopoietic stem/progenitor cells (HSPCs) and is associated with reduced mitochondrial function. These data show that heterozygous ATP-binding pocket variants in CLPB are a new and relatively common cause of congenital neutropenia.
Methods
Congenital neutropenia samples
Patients with congenital neutropenia were enrolled in the Severe Chronic Neutropenia International Registry (SCNIR) or the French Chronic Neutropenia Registry or identified through clinical sequencing. A total of 85 patients with congenital neutropenia was selected from the SCNIR based on prior ELANE genotyping, with the majority (70) having no ELANE mutation. Exome sequencing for the French registry cohort was performed on trios (proband-parents) after excluding the genes that are classically involved in SCN by targeted high-throughput sequencing. All patients gave informed consent for these studies under protocols approved by local institutional review boards. DNA was extracted from blood, bone marrow, or saliva samples.
Exome sequencing
Library preparation, sequencing, and data analysis details are provided in supplemental Materials and methods (available on the Blood Web site). Data were aligned to genome build GRCh38, and variants passing quality filters and present in the Exome Aggregation Consortium database15 at a frequency <1% were identified. Mean gene expression was derived from data using 3 healthy donors with populations defined as previously reported.16 The following criteria were used to identify potentially pathogenic variants: (1) variants that altered amino acid sequence, including missense, non-sense, or splice site variants, (2) missense variants predicted to be deleterious based on a combined annotation dependent depletion score ≥15, (3) variants with a frequency <0.0025 in the Exome Aggregation Consortium database,15 and (4) variants in genes that are highly expressed in granulocyte precursors (transcripts per million >2). Variants of interest were narrowed to those that had agreement between in silico predictions algorithms, as further outlined in supplemental Materials and methods. Copy number variation analysis was performed with cnvkit17 using default parameters to generate a reference copy number profile across all samples, followed by the ‘batch’ command for identification of copy number variation in each sample.
Human HSPC isolation and culture
Human umbilical cord blood was obtained from the Saint Louis Cord Blood Bank. Mononuclear cells were enriched using Lymphoprep (STEMCELL Technologies). CD34+ cells were isolated using biotin anti-CD34 (BioLegend) antibody, followed by enrichment with antibiotin MicroBeads (Miltenyi Biotec). CD34+ cells were resuspended in StemSpan SFEM II with 100 ng/mL of human stem cell factor (SCF), human thrombopoietin, and human Fms-like tyrosine kinase 3-ligand (PeproTech). Granulocytic differentiation was assessed by culturing cells in StemSpan SFEM II media with 3 ng/mL granulocyte-colony stimulating factor (G-CSF), 10 ng/mL of SCF, and 10% fetal calf serum for 10-14 days. Granulocyte colony-forming unit (CFU-G) assays were performed using MethoCult H4230 (STEMCELL Technologies) supplemented with G-CSF and SCF.
CRISPR/Cas9 knockout and lentiviral overexpression
A lentiviral vector was constructed based on the MND promoter for high expression in human hematopoietic cells.18,19 Human CLPB encoding isoform 2 (the highest expressed isoform in HSPCs and myeloid lineage cells) with a C-terminal hemagglutinin (HA) or c-Myc epitope tag was followed by an internal ribosomal entry site linked to green fluorescent protein (GFP) or blue fluorescent protein (BFP). Following HSPC enrichment, CD34+ cells were transduced with lentivirus at a multiplicity of infection of 15. To assess the degree of CLPB overexpression, RNA was isolated from day-7 cultures using a Spin-Column Purification Kit (MACHEREY-NAGEL), and complementary DNA (cDNA) was prepared using a Reverse Synthesis Kit (Bio-Rad). An exon-spanning probe was used to detect all isoforms of CLPB, and quantitative real-time polymerase chain reaction was performed using a TaqMan Universal PCR MasterMix Kit (Agilent Biosystems). For generation of knockout cells using the clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 system, single-guide RNA (sgRNAs) were designed using the Broad Institute Genetic Perturbation Platform CRISPRko Web-based tool (https://portals.broadinstitute.org/gpp/public/analysis-tools/sgrna-design). sgRNAs were ordered from Synthego with 2′-O-methyl 3′ phosphorothioate modifications in the capping nucleotides for increased stability. Recombinant Cas9 protein (IDT) was mixed with sgRNAs and incubated at room temperature to generate ribonuclear protein complexes, followed by nucleofection into HSPCs using the Neon system, as previously described.20 Insertion/deletion status was assessed by next-generation sequencing on the Illumina MiSeq platform and analyzed using Crispresso2.21
Flow cytometry
Cells were collected on the indicated day of culture, resuspended in phosphate-buffered saline (PBS) containing 0.1% bovine serum albumin with human Fc-block reagent (Human Trustain FcX; BioLegend), and incubated with antibodies on ice for 20 minutes. After washing, cells were resuspended in SYTOX Green (Invitrogen) for live/dead discrimination. Granulocyte precursors were defined as CD33+CD14−CD11b+/−CD16−, and mature neutrophils were defined as CD33+CD14−CD11b+CD16+. Data were collected on an Attune NxT Flow Cytometer and analyzed using FlowJo v10. For apoptosis assays on primary human HSPCs, cells were placed in serum-free media for 16 hours, stained with neutrophil differentiation markers, and fixed with BD Cytofix/Cytoperm solution (BD Biosciences). Cells were stained with anti-cleaved caspase 3 antibody (BioLegend). Apoptosis assays in the myeloid cell line MOLM-13 followed 24 hours of treatment with control dimethyl sulfoxide (DMSO) or the glycosylation-inhibiting drug tunicamycin (Sigma, St Louis, MO), which was resuspended in DMSO and used at a final concentration of 1 μM. Cells were washed in EDTA-free media and resuspended in Annexin V-PE, according to the manufacturer’s instructions, or were fixed and stained for cleaved caspase-3, as above. For cell cycle analysis, following fixation cells were stained with anti-Ki67 antibody, washed, and resuspended in FxCycle Violet (Invitrogen).
ATPase and disaggregase assays
Human CLPB protein containing aa 127-707, representing the ankyrin-rich repeat domains, ATPase domain, and C-terminal domain but lacking the N-terminal mitochondrial localization sequence and PARL-cleaved regulatory domains, was purified as previously described.11,22 ATPase activity was assessed via detection of inorganic phosphate release using a malachite green detection assay (Expedeon) measured on a Tecan Infinite M1000 or Safire 2 plate reader, as previously described.11,23 Values represent background (time 0) subtracted from end point colorimetric change. Luciferase disaggregation was performed using urea-denatured firefly luciferase, as previously described.11 Detection of recovered luminescence was monitored using the above plate readers. For the mixing studies, wild-type (WT) CLPB was mixed with WT or variant CLPB at equimolar ratios for 10 minutes at 25°C (total protein concentration 4 μM).
Confocal imaging
Following lentiviral transduction, MOLM-13 cells were cytospun onto glass slides, fixed using BD Cytoperm/Cytofix reagent, permeabilized with 0.5% Triton X-100 (Sigma), and stained with antibodies to detect the C-terminal HA epitope tag on CLPB (Cell Signaling Technology) or the mitochondrial marker TOM-20 (Cell Signaling Technology), followed by incubation with a secondary antibody (anti-Rabbit Alexa Fluor 594 or anti-Mouse Alexa Fluor 647). Nuclei were counterstained with SYTOX Green, and slides were mounted using ProLong Glass Antifade (Thermo Fisher) and imaged on an LSM 700 confocal microscope (Carl Zeiss Microscopy) using a 63× objective. Images were processed using Zeiss Zen software.
Mitochondrial assays
MOLM-13 cells were lentivirally transduced as above using a BFP reporter plasmid and sorted on BFP+ cells. The mitochondrial stress test was performed according to the manufacturer’s instructions and analyzed on a Seahorse XFe96 Analyzer (Agilent). Cells were then stained with Hoechst live cell nuclear stain (Sigma) and imaged on a Cytation 5 analyzer (BioTek). Seahorse data were normalized to cell number and processed using Wave software. To estimate mitochondrial mass, cells were washed with prewarmed PBS and stained with MitoTracker Green (Invitrogen) for 30 minutes in a 37°C incubator. Cells were then seeded on a Cell-Tak–coated 96-well black walled plate and imaged on a Cytation 5 analyzer. To estimate mitochondrial membrane potential, cells were washed with prewarmed PBS and adhered to a Cell-Tak–coated 96-well black walled plate using centrifugation and stained with the mitochondrial membrane potential sensitive dye tetramethylrhodamine methyl ester (TMRM; Sigma) for 20 minutes in a 37°C incubator. Cells were imaged on a Cytation 5 analyzer. Data were processed using Gen software (BioTek) to create a cell-size mask, within which mean fluorescence intensity was calculated.
Statistical analysis
Significance was determined using Prism v8.1.2 (GraphPad, San Diego, CA). For single-parameter analysis, an unpaired Student t test was used to assess statistical significance. For multiple-parameter data, statistical significance was calculated using 1- or 2-way analysis of variance (ANOVA). Values of P < .05 were considered significant.
Results
Heterozygous CLPB variants in congenital neutropenia
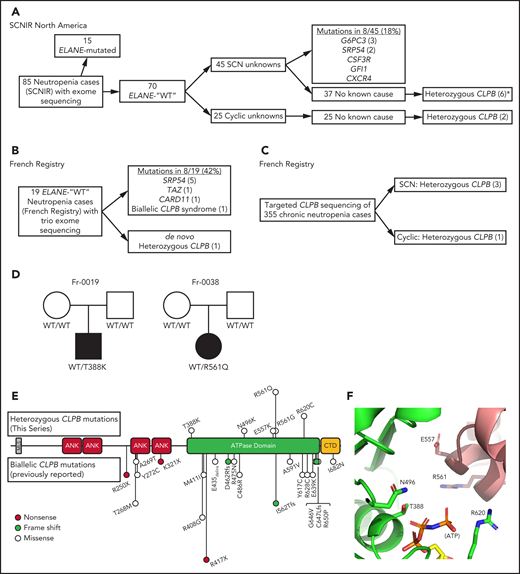
In an effort to identify novel genetic causes of SCN, we performed exome sequencing on 85 persons with congenital neutropenia recruited through the SCNIR; prior ELANE genotyping identified variants in 15 of 85 persons (supplemental Table 1). Using our filtering strategy defined in “Methods,” we identified all known cases of ELANE-mutated congenital neutropenia and 8 cases of SCN with variants in established SCN-associated genes (Figure 1A). In the remaining 62 cases of congenital neutropenia with no known genetic cause, we identified 4 unique heterozygous missense variants in CLPB in 5 persons with SCN. One additional SCN patient was initially identified through clinical sequencing and subsequently underwent exome sequencing, confirming their heterozygous CLPB variant and the absence of other known SCN causative gene variants. Independently, exome sequencing was performed on 19 persons (and their parents) with chronic neutropenia without a known genetic cause who were enrolled in the French Chronic Neutropenia Registry (Figure 1B). This identified 1 SCN patient with a de novo heterozygous CLPB variant. An additional 3 SCN cases with heterozygous CLPB variants were identified through targeted sequencing of 355 chronic neutropenia cases (Figure 1C). Altogether, we identified 6 unique CLPB variants within 10 unrelated SCN patients (Table 1). Most of these variants were confirmed by Sanger sequencing (supplemental Figure 1). For 2 of these patients, family studies were available and indicated a de novo inheritance pattern (Figure 1D). We also identified heterozygous CLPB variants in 2 cases of cyclic neutropenia in the SCNIR cohort, with a third case identified in the French Neutropenia Registry; interestingly, all 3 cases carried the CLPB R628C variant (supplemental Table 2). No copy number alterations in CLPB were detected. Of note, we identified 2 additional CLPB variants that passed our filtering strategy (R327W and R603H); however, both were present in an asymptomatic parent, indicating that they are likely benign and, therefore, are not included in Table 1.
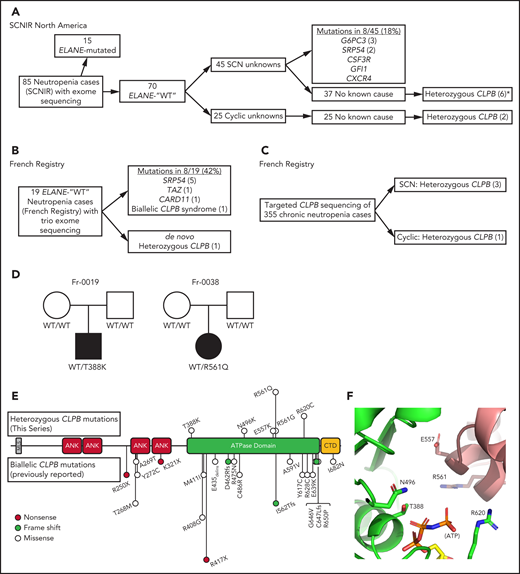
CLPB variants in SCN. Flow diagrams summarizing independent sequencing results from the SCNIR (A) and French SCN registry (B-C). The number of cases carrying a specific variant is shown in parentheses. (D) Pedigrees for 2 de novo cases with available family studies. Parental and patient variant status was confirmed using Sanger sequencing. WT indicates that the relevant CLPB variant was not present. Male (square) and female (circle) family members are depicted as unaffected (white) or affected (black). Parentage for cases Fr-0019 and Fr-0038 was confirmed using short tandem repeat analysis. (E) Mutational spectrum of CLPB as identified in this series (top) vs previously reported biallelic variants (bottom). The overall domain architecture of human CLPB is shown and includes the mitochondrial localization sequence (MLS), ankyrin-rich repeats (ANK), ATPase domain, and C-terminal domain (CTD). (F) CLPB protein structure model based on data from the crystal structure of T thermophilus CLPB (PDB ID: 1qvr). Side chains are shown for the 5 mutated residues, found in 10 patients, that cluster around the ATP-binding pocket. *One patient was identified initially through clinical sequencing but subsequently enrolled in the SCNIR and underwent exome sequencing. Cyclic, cyclic neutropenia.
CLPB variants in SCN. Flow diagrams summarizing independent sequencing results from the SCNIR (A) and French SCN registry (B-C). The number of cases carrying a specific variant is shown in parentheses. (D) Pedigrees for 2 de novo cases with available family studies. Parental and patient variant status was confirmed using Sanger sequencing. WT indicates that the relevant CLPB variant was not present. Male (square) and female (circle) family members are depicted as unaffected (white) or affected (black). Parentage for cases Fr-0019 and Fr-0038 was confirmed using short tandem repeat analysis. (E) Mutational spectrum of CLPB as identified in this series (top) vs previously reported biallelic variants (bottom). The overall domain architecture of human CLPB is shown and includes the mitochondrial localization sequence (MLS), ankyrin-rich repeats (ANK), ATPase domain, and C-terminal domain (CTD). (F) CLPB protein structure model based on data from the crystal structure of T thermophilus CLPB (PDB ID: 1qvr). Side chains are shown for the 5 mutated residues, found in 10 patients, that cluster around the ATP-binding pocket. *One patient was identified initially through clinical sequencing but subsequently enrolled in the SCNIR and underwent exome sequencing. Cyclic, cyclic neutropenia.
Prior studies showed that biallelic variants of CLPB are associated with a syndrome (CLPB syndrome) that is characterized by 3-MGA, cataracts, neurologic disease, and variable neutropenia.2-5 However, the variants seen in CLPB syndrome and CLPB-SCN are distinct. In patients with CLPB syndrome, the CLPB variants are always biallelic and are found scattered throughout the protein,6 with half of patients having ≥1 frameshift or non-sense variant (Figure 1E). In contrast, the variants observed in our series are heterozygous, missense, and localize to the C-terminal ATP-binding domain. We generated a structural model of human CLPB by threading the primary amino acid sequence onto that of Thermus thermophilus ClpB.24 All 6 of the heterozygous CLPB-SCN variants are predicted to contribute to the ATP-binding pocket (Figure 1E). Of note, these variants are in evolutionarily conserved residues and, by homology, most are predicted to be crucial residues for nucleotide binding and hydrolysis24-26 (supplemental Figure 2).
The clinical characteristics of the 10 patients with SCN carrying ATP-binding pocket CLPB variants are summarized in Table 1. All of the CLPB-SCN patients were diagnosed before the age of 5 years, and most received G-CSF therapy, with a median dose of 5.62 μg/kg per day (a typical dose for SCN treatment). All demonstrated a myeloid maturation arrest, and most had documented severe infections prior to G-CSF therapy. One patient developed a myeloid malignancy and is the only deceased patient from the cohort. In contrast to CLPB syndrome, in which 3-MGA is universal, none of the 5 patients in our series with available urine samples had 3-MGA.6 Likewise, although cataracts and neurologic abnormalities were present in >90% of cases of CLPB syndrome (very often co-occurring), they were uncommon in our CLPB-SCN cohort (2 nonoverlapping patients each with cataracts or epilepsy and 1 with developmental concerns). No case had >1 nonneutropenia CLPB syndrome feature. Of note, nearly 20% of patients with CLPB syndrome did not have neutropenia. The different clinical and molecular features of CLPB syndrome and CLPB-SCN suggest that these are distinct, but related, disorders with potentially unique mechanisms of disease pathogenesis.
Loss of CLPB results in impaired granulocytic differentiation
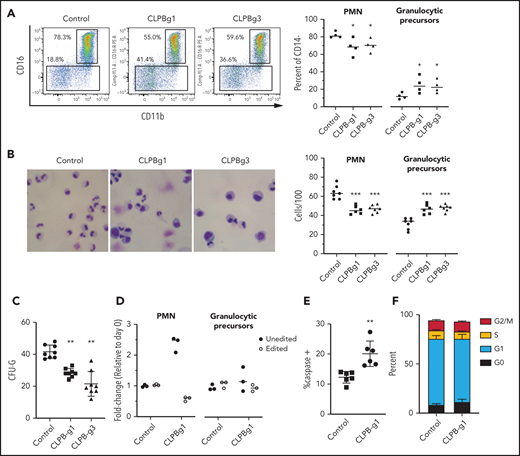
To assess the contribution of CLPB to granulopoiesis, we first used CRISPR/Cas9 gene editing to generate null mutations in CLPB in human cord blood CD34+ HSPCs (supplemental Figure 3A). We were able to achieve >80% editing efficiency, with a concordant decrease in protein expression (supplemental Figure 3B-D). The gene-edited HSPCs were cultured in the presence of G-CSF and SCF, and differentiation was assessed on day 14 by flow cytometry or by histomorphometry (Figure 2A-B). Compared with HSPCs transduced with control sgRNA, significantly fewer mature neutrophils and an increase in granulocytic precursors were observed in cells transduced with 2 independent sgRNAs targeting CLPB. A significant decrease in CFU-G also was observed (Figure 2C). Gene editing with the CRISPR/Cas9 system predominantly generates small insertion/deletions, some of which are in-frame and may yield functional intact CLPB protein. Next-generation sequencing of cells from day-14 cultures showed a significant enrichment for nontargeted and in-frame CLPB sequences in neutrophils, but not in granulocytic precursors, consistent with a selective loss of CLPB-deficient cells during terminal granulocytic differentiation (Figure 2D). Impaired granulocytic differentiation was due, at least in part, to increased apoptosis of early granulocytic precursors (Figure 2E), with no change observed in cell cycle status (Figure 2F; supplemental Figure 4). Together, these data show that CLPB is required for normal granulocytic differentiation.
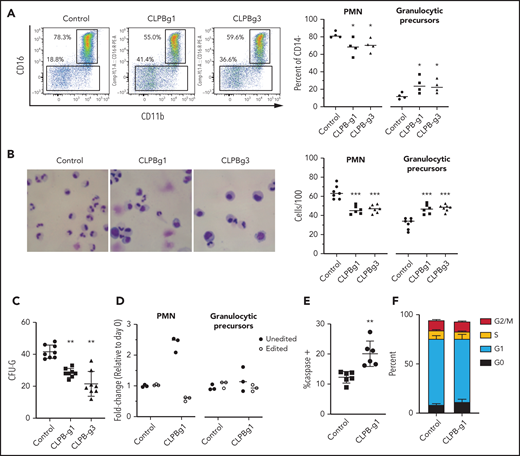
CLPB-deficient HSPCs have impaired granulocytic differentiation. Human cord blood CD34+ cells were nucleofected with guide RNA targeting CLPB (CLPB-g1 or CLPB-g3) or a control guide RNA targeting the intron of AAVS1 (as a gene editing control) complexed together with recombinant Cas9 protein. Edited CD34+ cells were cultured for 7-14 days in media containing G-CSF and SCF. (A) Representative flow plots showing gating strategy to identify CD11b+CD16+ mature neutrophils (PMN) or CD11b+/−CD16− granulocytic precursors; data are gated on CD14− cells to remove monocytes. Data are quantified in the right panel. (B) Representative hematoxylin/eosin-stained cytospin preparations of cells on day 14 of culture; original magnification using a 63X objective. Data are quantified in the right panel. (C) Number of CFU-G per 2000 gene-edited CD34+ cells. (D) Fold change (from day 0) in edited cells (for CLPB, edited out-of-frame) or unedited cells (for CLPB, unedited plus edited in-frame) in PMNs or granulocytic precursors sorted on day 14. (E) Percentage of caspase-3+ granulocytic precursors on day 7 of culture. (F) Cells were cultured for 7 days, and cell cycle was assessed by flow cytometry. Data represent 3-5 independent experiments. *P < .05, **P < .01, ***P < .005, repeated measures 1-way ANOVA.
CLPB-deficient HSPCs have impaired granulocytic differentiation. Human cord blood CD34+ cells were nucleofected with guide RNA targeting CLPB (CLPB-g1 or CLPB-g3) or a control guide RNA targeting the intron of AAVS1 (as a gene editing control) complexed together with recombinant Cas9 protein. Edited CD34+ cells were cultured for 7-14 days in media containing G-CSF and SCF. (A) Representative flow plots showing gating strategy to identify CD11b+CD16+ mature neutrophils (PMN) or CD11b+/−CD16− granulocytic precursors; data are gated on CD14− cells to remove monocytes. Data are quantified in the right panel. (B) Representative hematoxylin/eosin-stained cytospin preparations of cells on day 14 of culture; original magnification using a 63X objective. Data are quantified in the right panel. (C) Number of CFU-G per 2000 gene-edited CD34+ cells. (D) Fold change (from day 0) in edited cells (for CLPB, edited out-of-frame) or unedited cells (for CLPB, unedited plus edited in-frame) in PMNs or granulocytic precursors sorted on day 14. (E) Percentage of caspase-3+ granulocytic precursors on day 7 of culture. (F) Cells were cultured for 7 days, and cell cycle was assessed by flow cytometry. Data represent 3-5 independent experiments. *P < .05, **P < .01, ***P < .005, repeated measures 1-way ANOVA.
Expression of ATP-binding pocket CLPB variants results in impaired granulocytic differentiation
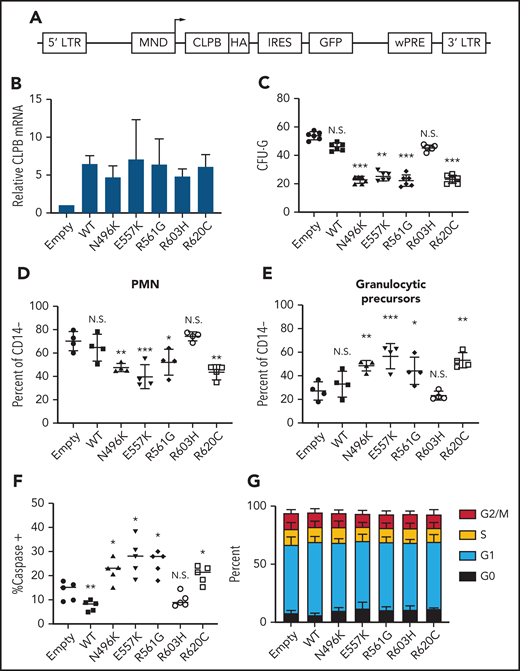
To assess the impact of heterozygous CLPB variants on granulopoiesis, we transduced cord blood CD34+ cells with lentivirus expressing each of the 4 CLPB variants from our SCNIR exome sequencing cohort (Figure 3A). We included the likely benign paternally inherited non-ATP binding cleft R603H variant as a negative control. Of note, because CLPB expression was increased fourfold to 10-fold compared with vector-alone controls (Figure 3B; supplemental Figure 5), we included a WT CLPB lentiviral cohort to control for the effect of CLPB overexpression (Figure 3B). A significant decrease in CFU-G was observed for the 4 CLPB variants that localize to the ATP-binding pocket (N496K, E557K, R561G, and R620C) but not for the R603H variant (Figures 1E and 3C). Fewer mature neutrophils and an increase in granulocytic precursors were present in cultures of HSPCs transduced with the ATP-binding pocket CLPB variants but not the R603H variant (Figure 3D-E). Expression of ATP-binding pocket variants resulted in an increase in apoptosis of granulocytic precursors but no change in cell cycle status (Figure 3F-G). Collectively, these data show that heterozygous ATP-binding pocket variants of CLPB cause impaired granulocyte differentiation.
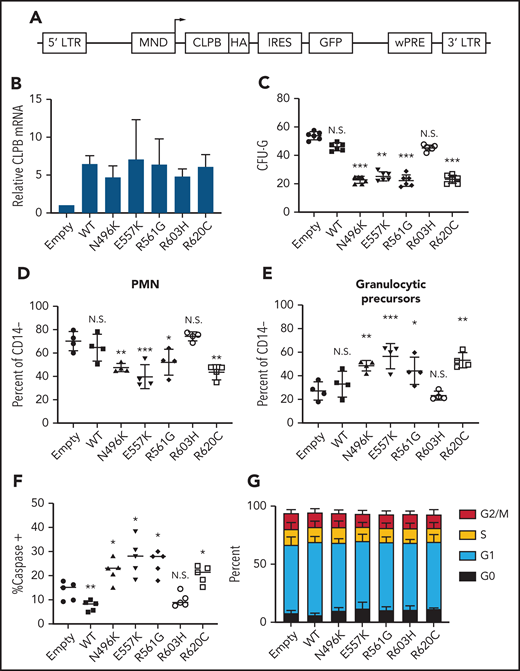
Expression of ATP-binding pocket CLPB mutants impairs granulocytic differentiation. Cord blood CD34+ cells were transduced with lentivirus expressing the indicated CLPB cDNA (or empty vector control). GFP+ cells were sorted at 48 hours and then seeded into media containing G-CSF and SCF or methylcellulose containing G-CSF. (A) Lentiviral vector. (B) RNA expression of CLPB relative to β-actin messenger RNA. (C) CFU-G per 2000 GFP+ CD34+ cells. Percentage of mature neutrophils (D) and granulocytic precursors (E) on day 14 of culture; data are gated on CD14− cells to exclude monocytes. (F) Percentage of caspase-3+ granulocytic precursors on day 7 of culture. (G) Cells were cultured for 7 days, and cell cycle was assessed by flow cytometry. Data represent 3-5 independent experiments. *P < .05, **P < .01, ***P < .005, repeated-measures 1-way ANOVA. IRES, internal ribosomal entry site; LTR, long terminal repeat; N.S., not significant; wPRE, woodchuck promoter responsive element.
Expression of ATP-binding pocket CLPB mutants impairs granulocytic differentiation. Cord blood CD34+ cells were transduced with lentivirus expressing the indicated CLPB cDNA (or empty vector control). GFP+ cells were sorted at 48 hours and then seeded into media containing G-CSF and SCF or methylcellulose containing G-CSF. (A) Lentiviral vector. (B) RNA expression of CLPB relative to β-actin messenger RNA. (C) CFU-G per 2000 GFP+ CD34+ cells. Percentage of mature neutrophils (D) and granulocytic precursors (E) on day 14 of culture; data are gated on CD14− cells to exclude monocytes. (F) Percentage of caspase-3+ granulocytic precursors on day 7 of culture. (G) Cells were cultured for 7 days, and cell cycle was assessed by flow cytometry. Data represent 3-5 independent experiments. *P < .05, **P < .01, ***P < .005, repeated-measures 1-way ANOVA. IRES, internal ribosomal entry site; LTR, long terminal repeat; N.S., not significant; wPRE, woodchuck promoter responsive element.
CLPB heterozygous variants have impaired ATPase and disaggregase activity and exert a dominant-negative effect on WT CLPB
Human CLPB is an ATPase with potent disaggregase activity.11,12 Biallelic CLPB variants that impair ATP cleavage also show impaired disaggregase activity, although there also appear to be ATPase-intact variants with impaired disaggregase activity that occur through alternative mechanisms.11 The heterozygous nature of our CLPB-SCN variants suggests a dominant-negative effect on WT CLPB. To test this hypothesis, we performed mixing studies with recombinant WT and mutant CLPB proteins. As a control, we included the R408G CLPB variant that is found in biallelic CLPB syndrome in conjunction with a second CLPB variant. Of note, parents with heterozygous R408G CLPB are asymptomatic,2,6 even though purified R408G protein has impaired ATPase activity.2 We also included the likely benign variant (R603H). We show that all of the CLPB variants (with the exception of the R603H control) have impaired ATPase and disaggregase activity when tested in isolation (Figure 4A-B). When mixed with WT CLPB, all of the heterozygous CLPB-SCN variants demonstrate a >50% reduction in ATPase activity (reaching significance for E557K and R561G) and disaggregase activity (reaching significance for all CLPB-SCN variants) (Figure 4C-D). In contrast, the R408G variant, although having reduced ATPase and disaggregase activity in isolation (Figure 4A-B), did not suppress the activity of WT CLPB (Figure 4C-D). As expected, the R603H variant had no effect. Together, these data suggest that heterozygous CLPB-SCN variants act in a dominant-negative fashion.
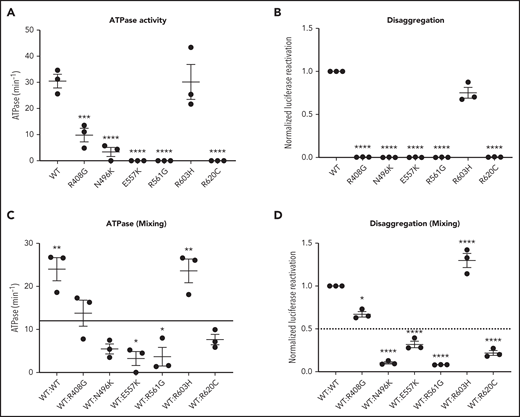
CLPB mutants show impaired ATPase and disaggregase activity and exhibit a dominant-negative effect on WT CLPB. Purified CLPB protein was used to measure ATPase activity of the indicated CLPB variants tested in isolation (A); disaggregase activity of the indicated CLPB variants tested in isolation (B); ATPase activity of WT CLPB when mixed 1:1 with WT CLPB or CLPB variants (C); and disaggregase activity of WT CLPB when mixed 1:1 with WT CLPB or CLPB variants (D). In (C-D), the dotted line represents 50% of WT CLPB activity. Data represent 3 independent experiments. *P < .05, **P < .01, ***P < .001, ****P < .0001, 1-way ANOVA. In (A-B), comparison was made with WT CLPB activity. A comparison was made with values representing 50% of WT activity (C-D).
CLPB mutants show impaired ATPase and disaggregase activity and exhibit a dominant-negative effect on WT CLPB. Purified CLPB protein was used to measure ATPase activity of the indicated CLPB variants tested in isolation (A); disaggregase activity of the indicated CLPB variants tested in isolation (B); ATPase activity of WT CLPB when mixed 1:1 with WT CLPB or CLPB variants (C); and disaggregase activity of WT CLPB when mixed 1:1 with WT CLPB or CLPB variants (D). In (C-D), the dotted line represents 50% of WT CLPB activity. Data represent 3 independent experiments. *P < .05, **P < .01, ***P < .001, ****P < .0001, 1-way ANOVA. In (A-B), comparison was made with WT CLPB activity. A comparison was made with values representing 50% of WT activity (C-D).
Expression of ATP-binding pocket CLPB variants results in impaired mitochondrial function
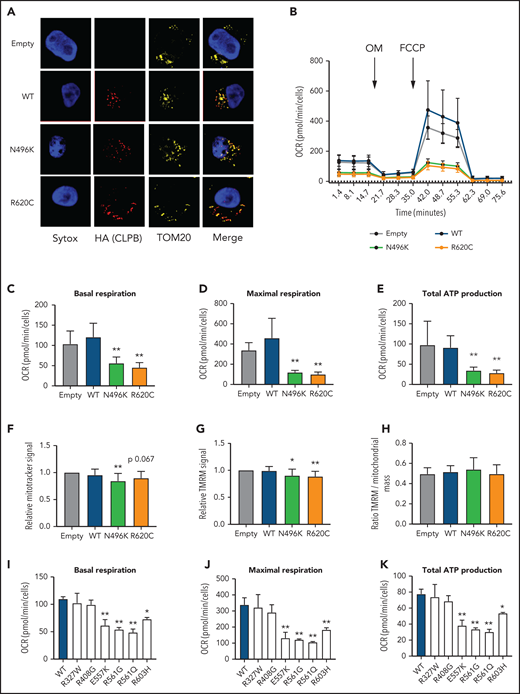
CLPB contains a mitochondrial localization sequence and has previously been shown to localize to the mitochondria in a human myeloid progenitor cell line (MOLM-13),14 and the absence of CLPB causes impaired mitochondrial protein solubility.11 Therefore, we hypothesized that CLPB-SCN heterozygous variants may affect granulocyte progenitors by impacting mitochondrial function. To test this hypothesis, we generated MOLM-13 cells expressing WT or mutant CLPB. We initially focused on CLPB N496K and R620C. WT and mutant CLPB localize to mitochondria (Figure 5A). CLPB mutant cells had impaired mitochondrial respiration, with significantly reduced basal and maximal respiratory capacity and a corresponding decrease in ATP production (Figure 5B-E). A small, but significant, decrease in mitochondrial mass was observed in CLPB-N496K–overexpressing cells, with a corresponding decrease in mitochondrial membrane potential, as measured using TMRM (Figure 5F-H).

Expression of CLPB mutants impairs mitochondrial respiration without affecting membrane potential. MOLM-13 cells were transduced with a lentiviral vector expressing WT CLPB, CLPB-N496K, or CLPB-R620C. (A) Representative photomicrographs of cells stained with anti-HA antibody to detect CLPB (red) and anti-TOM20 (yellow) to label mitochondria. Nuclei were counterstained with SYTOX Green (blue); original magnification, ×63. (B-E) The mitochondrial stress test was performed using a Seahorse XFe96 Analyzer. (B) Representative graph showing the oxygen consumption rate (OCR) at baseline and after treatment with the ATP synthase inhibitor oligomycin (OM) and then the uncoupling agent carbonyl cyanide-4 phenylhydrazone (FCCP). Parameters of mitochondrial respiration include basal respiration (C), maximal respiration (D), and total ATP production (E). (F-H) Cells were labeled TMRM or MitoTracker Green. (F) MitoTracker Green signal normalized to empty vector control. (G) TMRM signal normalized to empty vector control. (H) Ratio of TMRM signal/MitoTracker green signal. Mitochondrial respiration parameters for additional CLPB variants: basal respiration (I), maximal respiration (J), and total ATP production (K). *P < .05, **P < .01, repeated-measures 1-way ANOVA.
Expression of CLPB mutants impairs mitochondrial respiration without affecting membrane potential. MOLM-13 cells were transduced with a lentiviral vector expressing WT CLPB, CLPB-N496K, or CLPB-R620C. (A) Representative photomicrographs of cells stained with anti-HA antibody to detect CLPB (red) and anti-TOM20 (yellow) to label mitochondria. Nuclei were counterstained with SYTOX Green (blue); original magnification, ×63. (B-E) The mitochondrial stress test was performed using a Seahorse XFe96 Analyzer. (B) Representative graph showing the oxygen consumption rate (OCR) at baseline and after treatment with the ATP synthase inhibitor oligomycin (OM) and then the uncoupling agent carbonyl cyanide-4 phenylhydrazone (FCCP). Parameters of mitochondrial respiration include basal respiration (C), maximal respiration (D), and total ATP production (E). (F-H) Cells were labeled TMRM or MitoTracker Green. (F) MitoTracker Green signal normalized to empty vector control. (G) TMRM signal normalized to empty vector control. (H) Ratio of TMRM signal/MitoTracker green signal. Mitochondrial respiration parameters for additional CLPB variants: basal respiration (I), maximal respiration (J), and total ATP production (K). *P < .05, **P < .01, repeated-measures 1-way ANOVA.
We next expanded our analysis to include other ATP-binding pocket variants from our heterozygous CLPB-SCN cohort (E557K, R561G, R561Q) and the 2 parentally inherited likely benign variants (R327W and R603H). We also tested the R408G ATP-binding pocket variant that is found in CLPB syndrome patients, always in combination with non-sense or frameshift variants. As expected, the additionally tested ATP-binding pocket variants from our heterozygous-CLPB SCN cohort also demonstrated impaired mitochondrial function (Figure 5I-K). In contrast, 1 of the paternally inherited variants (R327W) did not. The other paternally inherited variant (R603H) displayed an intermediate phenotype. Interestingly, the R408G variant from the biallelic CLPB syndrome series did not show impaired mitochondrial function. The observed differences were not due to impaired association of mutant CLPB with WT CLPB, because R408G and our heterozygous CLPB-SCN variants were able to co-associate with WT CLPB (supplemental Figure 6). Together, these data show that expression of ATP-binding pocket CLPB-SCN variants results in impaired mitochondrial respiration.
Expression of ATP-binding pocket CLPB variants does not render MOLM-13 cells more sensitive to ER stress
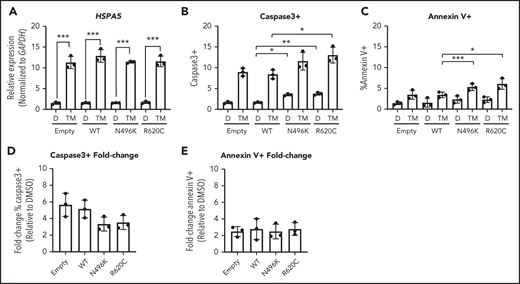
During granulocytic differentiation, the massive expression of neutrophil elastase (encoded by ELANE), and other primary granule proteins, induces endoplasmic reticulum (ER) stress. Indeed, we and other investigators reported that misfolded neutrophil elastase, through induction of ER stress, contributes to the pathogenesis of ELANE-mutated SCN.27-30 ER stress can induce the unfolded protein response (UPR), ultimately resulting in apoptosis. Recent studies suggest that UPR-induced calcium efflux can enhance mitochondrial respiration, resulting in protection from ER stress–induced apoptosis.31,32 Based on these observations, we hypothesized that the impaired mitochondrial respiration induced by mutant CLPB expression renders granulocytic precursors more sensitive to ER stress. To test this hypothesis, we induced ER stress in MOLM-13 cells expressing CLPB N496K or R620C by treating them with the glycosylation-inhibiting drug tunicamycin. The magnitude of ER stress induced by tunicamycin, as measured by HSPA5 messenger RNA expression, was similar in control and CLPB mutant cells (Figure 6A). In control (DMSO-treated) cells, expression of N496K and R620C results in a small, but significant, increase in apoptosis, as measured by cleaved caspase 3 (Figure 6B), with a trend toward increased surface expression of Annexin V (Figure 6C). Although there was a significant increase in the total percentage of apoptotic cells in the N496K and R620C groups, all variants showed an equivalent fold change increase in apoptosis relative to DMSO control (Figure 6D-E). Thus, mutant CLPB expression does not render cells more sensitive to ER stress–induced apoptosis.

Induction of UPR does not cause increased fold change in apoptosis in CLPB variant–expressing myeloid cells. The myeloid cell line MOLM-13 was transduced with CLPB variants or empty vector control, and cells were sorted based on GFP positivity. Cells were treated with DMSO (D) or the glycosylation-inhibitor tunicamycin (TM) for 24 hours. (A) Induction of the UPR was confirmed by assessing expression of HSPA5 by quantitative polymerase chain reaction. The percentage of cleaved caspase-3+ (B) or Annexin V+ (C) cells was assessed by flow cytometry. Fold change representation of cleaved caspase-3+ (D) or Annexin V+ (E) cells expressed as the ratio of TM/DMSO within each sample. Data represent 3 independent experiments. *P < .05, **P < .01, ***P < .001, repeated-measures 1-way ANOVA.
Induction of UPR does not cause increased fold change in apoptosis in CLPB variant–expressing myeloid cells. The myeloid cell line MOLM-13 was transduced with CLPB variants or empty vector control, and cells were sorted based on GFP positivity. Cells were treated with DMSO (D) or the glycosylation-inhibitor tunicamycin (TM) for 24 hours. (A) Induction of the UPR was confirmed by assessing expression of HSPA5 by quantitative polymerase chain reaction. The percentage of cleaved caspase-3+ (B) or Annexin V+ (C) cells was assessed by flow cytometry. Fold change representation of cleaved caspase-3+ (D) or Annexin V+ (E) cells expressed as the ratio of TM/DMSO within each sample. Data represent 3 independent experiments. *P < .05, **P < .01, ***P < .001, repeated-measures 1-way ANOVA.
Discussion
We identified 10 unrelated individuals with SCN and heterozygous ATP-binding pocket variants in CLPB. These patients present with isolated severe neutropenia with few, if any, of the nonhematopoietic features associated with biallelic CLPB loss, including 3-MGA. Expression of these CLPB variants in primary human HSPCs is sufficient to impair granulocytic differentiation. These observations suggest that heterozygous variants in CLPB that localize to the ATP-binding pocket are a new and relatively common cause of SCN. Indeed, in the North American exome sequencing cohort, potentially pathogenic heterozygous CLPB mutations were identified in 5 of 70 (7.1%) of ELANE-WT SCN cases, making it the second most common cause of SCN behind ELANE mutations. We also establish the presence of potentially pathogenic heterozygous CLPB variants in the European population.
Not all heterozygous mutations in CLPB will be disease causing. Our data support a model whereby highly conserved mutations located within the ATP-binding pocket are predicted to be pathogenic. Our combination of sequencing and functional data allows us to classify the variants in 10 of these individuals as pathogenic or likely pathogenic according to American College of Medical Genetics criteria33 (supplemental Table 3). We also identified 2 individuals with heterozygous variants in CLPB (R327W and R603H) that our functional data and inheritance patterns suggest are benign variants (supplemental Table 4). Of note, we also identified 3 unrelated families with cyclic neutropenia carrying the CLPB variant R628C. Although not functionally validated, these data raise the possibility that this heterozygous CLPB variant may be a rare cause of cyclic neutropenia. Together, these observations indicate that genetic testing for CLPB should be included in the work-up of patients presenting with congenital neutropenia and, importantly, that heterozygous mutations in the ATP-binding pocket should be considered potentially pathogenic.
The mechanisms by which heterozygous ATP-binding pocket CLPB variants impair granulocytic differentiation are not clear. Genetic and biochemical evidence support a dominant-negative mechanism. Parents of patients with CLPB syndrome carrying heterozygous-null CLPB mutations are asymptomatic.2-6 Additionally, we show that ATP-binding pocket CLPB mutants coimmunoprecipitate with WT CLPB and inhibit the ATPase and disaggregase activity of WT CLPB in mixing studies. On the other hand, a simple dominant-negative mechanism does not account for all of the phenotypic differences between persons with heterozygous CLPB mutations and CLPB syndrome. In particular, 3-MGA was not detected in the urine of all evaluable CLPB-SCN cases (5/10). In Pronicka et al’s review of 31 patients with CLPB,6 urine 3-MGA was detected in 29 of 29 evaluable cases, whereas it was not detected in any of the 5 cases of CLPB-SCN for which urine was available (P < .0001 vs 0/5 CLPB-SCN cases, Fisher’s exact test). These observations suggest the possibility that, although the ATPase activity of CLPB is needed to maintain mitochondrial functions required for granulocytic differentiation, an (undefined) ATPase-independent activity of CLPB helps to maintain mitochondrial functions required to suppress 3-methylglutaconic acid production. In this model, the biallelic CLPB mutations in CLPB syndrome patients results in a loss (or partial loss) of all CLPB functions, leading to neutropenia, 3-MGA, and other nonhematopoietic phenotypes. In contrast, the heterozygous ATP-binding pocket mutations of CLPB found in SCN cases only impair the ATPase-dependent functions of CLPB, leading to severe neutropenia without 3-MGA. Further study is needed to test this model, including careful examination for extrahematopoietic phenotypes in all identified heterozygous CLPB-SCN cases. This will allow for clarification of whether ATP-binding pocket CLPB mutations define a unique clinical entity or contribute to a wider clinical spectrum of CLPB deficiency.
Our data suggest that expression of ATP-binding pocket CLPB mutants results in impaired mitochondrial function and is similar to the phenotype of CLPB-null cells.14 Of note, our data show that mitochondrial respiration is significantly reduced in cells expressing mutant CLPB; this difference is not explained by the small (although significant) decrease in mitochondrial mass. This may be relevant, because during granulocytic differentiation there is a switch in energy metabolism from glycolysis to oxidative phosphorylation.34-36 Whether impaired oxidative phosphorylation contributes to the impaired granulocytic differentiation induced by mutant CLPB requires further study. Although recent studies have suggested a link between ER stress and mitochondrial stress, our data suggest that the mitochondrial dysfunction induced by mutant CLPB expression does not appear to render cells more sensitive to ER stress. CLPB protein interacts with the mitochondrial proteins optic atrophy 1 (OPA1) and HAX111,14 and is required for HAX1 solubility.11 Mutations of HAX1 are a rare cause of SCN,37 and OPA1 has been shown to regulate mitochondrial respiratory capacity through maintenance of mitochondrial cristae.38 Whether heterozygous ATP-binding pocket mutations of CLPB alter interactions with OPA1 or HAX1 to disrupt mitochondrial function requires further study. Interestingly, although neutropenia is observed in some mitochondrial syndromes, such as Barth syndrome, it is not a common feature.39 Thus, the specific mitochondrial defect may determine whether granulopoiesis is disrupted.
Establishing the link between SCN and heterozygous ATP-binding pocket mutations in CLPB will help to guide the clinical management of these patients: they should be treated with G-CSF therapy, as indicated, and monitored closely for infection and development of myeloid malignancy. Our findings also establish an important cell-intrinsic role for CLPB in normal human granulopoiesis. Future studies will be aimed at understanding the link between mitochondrial dysfunction and impaired granulocytic differentiation.
Acknowledgments
The authors are grateful to the persons who contributed samples and clinical information for this study. They also thank the Alvin J. Siteman Cancer Center at Washington University School of Medicine and Barnes-Jewish Hospital (St Louis, MO) for the use of the Siteman Flow Cytometry Core, which provided cell sorting expertise; Jessica Hoisington-Lopez and MariaLynn Cosby (DNA Sequencing Innovation Laboratory, Edison Family Center for Genome Sciences and Systems Biology) for expertise with DNA sequencing; Sridhar Nonavinkere Srivatsan for bioinformatics support; Severine Clauin for sequencing support; Julien Buratti for bioinformatics analysis support; James Huang for providing comments on the manuscript; and Paul Coppo, Mohamed Hamidou, and Amelie Servettaz for clinical expertise.
This work was supported by National Institutes of Health (NIH), Eunice Kennedy Shriver National Institute of Child Health and Human Development Institutional National Research Service Award Training Program in Developmental Hematology (T32 HD007499-19) and Training of the Pediatric Physician-Scientist (T32 HD043010), an American Society of Hematology Scholar Award, and Children’s Discovery Institute Fellowship MC-F-2020-871 (all to J.T.W.); NIH, National Institute on Aging grant F31 AG060672, NIH, National Institute of General Medical Sciences grant T32 GM008275, and a Blavatnik Family Foundation Fellowship (all to R.R.C.); NIH, National Cancer Institute grant K08 CA190815 (D.H.S.); The G. Harold and Leila Y. Mathers Foundation and NIH, National Institute of General Medical Sciences grant R01 GM099836 (both to J.S.); INSERM ITMO Sante publique, X4 Pharma, Prolong Pharma, and Chugai SA (French SCN Registry); Foundation for Rare Diseases (AO9102LS), 111 Les Arts, Association pour la Recherche et les Maladies Hématologiques de l'Enfant, Association de Barth, and the Association Sportive de Saint Quentin Fallavier (C.B.-C. and J.D.); NIH, National Institute of Allergy and Infectious Diseases grant 2R 24 AI 049393-Severe Chronic Neutropenia International Registry (D.C.D.); and US Department of Defense grant BM130173 and NIH, National Heart, Lung, and Blood Institute grant R01 HL152632-01 (both to D.C.L.).
Authorship
Contribution: D.C.L. and D.C.D. conceived and supervised the study; J.T.W., R.R.C., J.S., D.C.D., and D.C.L. designed the experiments; J.T.W., R.R.C., P.W., and N.L.K. performed the experiments; D.H.S. and A.E.L. performed sequence alignment and variant annotation; J.T.W., P.W., and D.C.L. analyzed SCNIR exome data and performed filtering; D.C.D., V.M., M.L.K., and A.A.B. provided patient samples and clinical information from the SCNIR; C.B.-C. and J.D. provided sequencing and clinical information from the French SCN Registry; J.T.W. and D.C.L. wrote the manuscript; and all authors reviewed and contributed to the final version of the manuscript.
Conflict-of-interest disclosure: J.S. is a consultant for Dewpoint Therapeutics and Maze Therapeutics. The remaining authors declare no competing financial interests.
Correspondence: Daniel C. Link, Department of Medicine, 660 South Euclid Ave, Box 8007, St Louis, MO 63110; e-mail: danielclink@wustl.edu; and David C. Dale, Department of Medicine, University of Washington, Box 356422, Seattle, WA 98195; e-mail: dcdale@uw.edu.
Data sharing requests should be sent to Daniel C. Link (danielclink@wustl.edu) or David C. Dale (dcdale@uw.edu).
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.