

In this issue of Blood, 1 characterize a novel Bruton tyrosine kinase (BTK) inhibitor, vecabrutinib, that targets mutant BTK C481S and wild-type BTK in preclinical models. This is the first BTK inhibitor that also inhibits interleukin-2–inducible T-cell kinase (ITK) and can still bind to mutant BTK.
Clinical outcomes in chronic lymphocytic leukemia (CLL) have greatly improved over the last 2 decades with the development of regimens combining cytotoxic drugs with anti-CD20 monoclonal antibodies,2 but outcomes vary significantly. For instance, patients in genetic high-risk groups, such as those harboring TP53 mutations, continue to have suboptimal outcomes despite treatment with the best available therapy.3 Similarly, patients who relapse early or are refractory to chemotherapy have an unfavorable outcome.4 These areas of ongoing unmet clinical need lend impetus to developing truly novel targeted approaches to treating high-risk B-cell leukemias. Among the most promising classes of targeted therapies in CLL are inhibitors of BTK (BTKi’s). BTK plays a prominent role in the BCR signaling pathway. Clinical activity of ibrutinib (the first US Food and Drug Administration–approved BTKi) is attributed to attenuated homing and retention of CLL cells to the microenvironment as a result of impaired BCR-controlled integrin-mediated adhesion and chemokine-controlled migration.5
Microenvironmental crosstalk plays an important role in CLL pathogenesis and progression. CLL cells are strongly dependent on interactions with other immune cells, thus shaping a highly orchestrated network: the tumor microenvironment.6 The inhibitory effects of ibrutinib on microenvironment homing and adhesion correlate with its clinical efficacy because ibrutinib treatment causes a rapid reduction of the lymph node size followed by a prolonged lymphocytosis.7 This prolonged lymphocytosis resulting from treatment with kinase inhibitors seems to have no clinical disadvantage.7 However, it could enhance the possibility of resistant clones accumulating. Acquired resistance to ibrutinib was reported in ∼80% of patients as a result of mutations in BTK itself or the downstream kinase phospholipase Cγ2 (PLCγ2).8 The BTK C481, most commonly serine, mutation confers resistance by preventing the covalent binding of ibrutinib to its target cysteine 481 in BTK (C481S).8
To date, 5 noncovalent BTKi’s that can inhibit the kinase in the presence of a BTK C481 mutation9 have entered clinical trials. In their article, Jebaraj et al characterized the noncovalent BTKi vecabrutinib (SNS-062) and demonstrated binding in the adenosine triphosphate binding pocket of BTK independent of the C481 residue (see figure). Vecabrutinib inhibited BCR signaling in wild-type and BTK C481S–mutant cells as measured by calcium flux. Furthermore, adoptive Eμ-TCL1 mice treated with vecabrutinib have increased survival compared with vehicle control (median survival, 35 vs 28 days). Interestingly, like ibrutinib, vecabrutinib is also a potent ITK inhibitor (50% inhibitory concentration,14 nM). ITK is downstream of the T-cell receptor. The T cells present in the tumor microenvironment function as supporters of CLL, and it has also become clear that CLL cells actively recruit supportive regulatory T cells.6 It has been reported that ibrutinib can reverse defects in T cells.10 So the authors investigated whether vecabrutinib, like ibrutinib, has immunomodulatory effects. Treatment with either ibrutinib or vecabrutinib reduced the number of immunosuppressive CD4+ regulatory T cells in Eμ-TCL1 mice, which suggests that vecabrutinib can reduce the supportive functions found in the microenvironment (see figure).
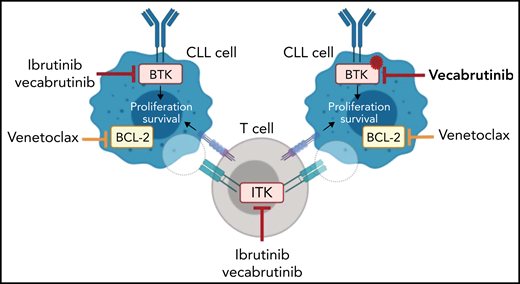
The noncovalent BTK/ITK inhibitor vecabrutinib can block wild-type and C481S-mutant BTK in preclinical models. Vecabrutinib has immunomodulatory effects similar to those of ibrutinib in the murine Eμ-TCL1 model. Treatment with a combination of vecabrutinib and venetoclax leads to prolonged survival of Eμ-TCL1 mice. Illustration created with BioRender.com.
The noncovalent BTK/ITK inhibitor vecabrutinib can block wild-type and C481S-mutant BTK in preclinical models. Vecabrutinib has immunomodulatory effects similar to those of ibrutinib in the murine Eμ-TCL1 model. Treatment with a combination of vecabrutinib and venetoclax leads to prolonged survival of Eμ-TCL1 mice. Illustration created with BioRender.com.
Because ibrutinib and vecabrutinib showed only a limited cytotoxic effect in CLL cells, combination with a drug that induces rapid apoptosis would be favorable. Venetoclax directly targets BCL-2 (a key regulator of programmed cell death) and is highly expressed in CLL cells. Jabaraj et al demonstrated that vecabrutinib, similar to ibrutinib, primes CLL cells to BCL-2 dependency (see figure). Subsequently, treatment with the combination of vecabrutinib and venetoclax resulted in prolonged survival of Eμ-TCL1 mice.
Approved BTKi-based regimens combined with BCL-2 inhibitor–based regimens are now well advanced in clinical trials,9 and evaluation is needed to determine whether such combination therapies reduce the development of BTK and PLCγ2-mutated clones. Combining vecabrutinib with venetoclax could overcome the hurdle of development of BTK mutations and hopefully achieve long-term remissions, an essential step toward improving outcomes for patients with CLL.
Conflict-of-interest disclosure: R.T. is an employee of the Walter and Eliza Hall Institute, which receives milestone and royalty payments related to venetoclax.

