

In this issue of Blood, Goswami et al1 report a new role for the tumor suppressor protein phosphatase 2A (PP2A) in regulating differentiation and growth arrest in acute myeloid leukemia (AML). This finding is important as PP2A inhibition is common in AML, yet how PP2A inhibition contributes to AML pathogenesis is not well defined. Importantly, several PP2A activating drugs are in development, and this study highlights the therapeutic potential of one of these, OSU-2S, revealing inhibition of leukemic-initiating cells and induction of terminal differentiation (see figure).
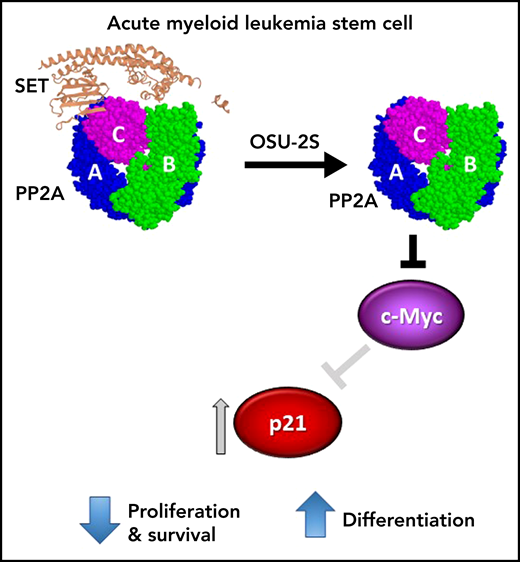
AML stem cell. OSU-2S binds to SET (PDB: 2E50), displacing it from the protein phosphatase 2A (PP2A) heterotrimer (PDB: 2IAE), thus increasing phosphatase activity. In AML stem cells, activated PP2A results in reduced expression of cMYC and subsequent induction of p21, leading to inhibition of leukemia-initiating cell proliferation and survival and terminal myeloid differentiation.
AML stem cell. OSU-2S binds to SET (PDB: 2E50), displacing it from the protein phosphatase 2A (PP2A) heterotrimer (PDB: 2IAE), thus increasing phosphatase activity. In AML stem cells, activated PP2A results in reduced expression of cMYC and subsequent induction of p21, leading to inhibition of leukemia-initiating cell proliferation and survival and terminal myeloid differentiation.
PP2A is a family of serine/threonine phosphatases that negatively regulate numerous oncogenic signaling pathways and often function as tumor suppressors. While inactivated in many cancers, PP2A is rarely mutated and thus is an attractive target of small molecule activators.2 PP2A is a multimeric enzyme composed of a catalytic subunit (C), a structural subunit (A), and a variable regulatory subunit (B), of which there are 4 different families, each with several isoforms. The association of a particular B subunit with the core AC dimer introduces substrate specificity and subcellular targeting of PP2A activity. PP2A activity is further regulated by post-translational modifications and by endogenous inhibitory proteins such as SET, CIP2A, SETBP1, and SBDS, which are often overexpressed in cancers, including AML.2,3
PP2A activity is frequently reduced in myeloid leukemias. In AML, this is often associated with oncogenic mutations in receptor tyrosine kinases FLT3 and cKIT, where low PP2A activity enhances the activation of FLT3- and cKIT-mediated signaling pathways and is essential for leukemia cell growth and survival.4-7 Goswami et al1 now demonstrate that PP2A inhibition is also essential for AML stem cell survival and is a key mediator of myeloid differentiation, with PP2A inhibition contributing to the characteristic differentiation block observed in AML.
The authors investigated the effects of pharmacological PP2A activation in AML cell lines, primary AML patient samples, and mouse models. There are 2 distinct classes of small molecule PP2A activators: sphingosine analogs, which activate PP2A by displacing PP2A inhibitory proteins; and tricyclic sulfonamides (or small molecule activators of PP2A [SMAPs]), which allosterically activate PP2A. The sphingosine analogs are derivatives of FTY720, which shows efficacy in preclinical cancer studies, including in AML. However, FTY720 is immunosuppressive on account of its phosphorylation by sphingosine kinase, binding to sphingosine 1-phosphate receptors, and inhibition of lymphocyte trafficking. Thus, sphingosine analogs that cannot be phosphorylated, such as OSU-2S,1 AAL(S),6 or CM-1231,8 are preferred compounds for clinical development. A key feature of pharmacological PP2A activation is the simultaneous inhibition of multiple kinase pathways such as PI3K and MAPK pathways; inhibition of transcription factors and oncogenic effectors such as MYC and RNAPII; and inhibition of antiapoptotic regulators such as BCL2 and MCL1. These multiple attributes may prevent or overcome treatment resistance that is commonly observed in targeted kinase inhibitor therapy.9 Goswami’s1 finding that pharmacological activation of PP2A drives maturation of AML cells and inhibits leukemia-initiating stem cells provides strong support for the clinical development of these compounds.
Goswami et al1 focus on OSU-2S, which they show activates PP2A by disrupting the association of PP2A with the inhibitory protein SET. OSU-2S was cytotoxic to AML cell lines and primary AML cells both in monoculture and when cocultured with bone marrow (BM) stromal cells, which is important as the BM microenvironment often protects AML cells from therapeutic targeting. FLT3- and cKIT-mutant AML has previously been shown to be sensitive to PP2A activators.4-6 Excitingly here, however, there was no clear association of OSU-2S sensitivity with mutations or FAB subtypes, suggesting a broad applicability for this approach in AML therapy.
Mechanistically, the OSU-2S-induced differentiation was blocked by molecular knockdown of PP2A catalytic subunit or the B56α regulatory subunit, suggesting a specific role for B56α. Differentiation was accompanied by cell cycle arrest at S-phase entry, reduced c-Myc protein expression, induction of p21, and dephosphorylation of the cell cycle mediator, retinoblastoma protein (Rb). Importantly, p21 knockdown rescued the OSU-2S-induced dephosphorylation of Rb, differentiation, and cell death; while overexpression of c-Myc rescued the OSU-2S-mediated induction of p21, cell death, and differentiation (see figure). Together this suggests that PP2A-mediated p21 upregulation is dependent on c-Myc downregulation. Targeting c-Myc has remained an elusive goal in anticancer therapy, but degradation of c-Myc induced by PP2A activators is an attractive approach to achieve this.
The antileukemic and differentiation-inducing effects of PP2A activation were confirmed in vivo. OSU-2S treatment increased survival and drove leukemic cell maturation in mice bearing MV4-11 xenografts and in an immunocompetent mouse model incorporating adoptive transfer of leukemic cells from Tet2−/−Flt3ITD mice. Finally, OSU-2S was shown to inhibit human AML stem cells. Importantly, normal HSPCs were ∼7-fold less sensitive to OSU-2S, and there was no effect on normal immune cell reconstitution in mice, providing a therapeutic window for the approach.
An important aspect of this study to be commended was the analysis of multiple different PP2A complexes. Most studies measure PP2A activity by coimmunoprecipitation using an antibody targeted to the catalytic subunit. However, this approach favors isolation of B’’’ (striatin)-containing PP2A complexes over others.10 Using this assay, Goswami et al demonstrated robust OSU-2S-induced PP2A activation and displacement of SET. They further coimmunoprecipitated for B55α and B56α PP2A complexes, and importantly showed that the phosphatase activity of both complexes was increased by OSU-2S. SET is known to interact with B56 complexes; thus the increased activity in these complexes is likely mediated by SET displacement. Whether the increased activity of B55α complexes is also mediated by SET displacement, or other PP2A inhibitory proteins such as SBDS,3 remains to be determined. Nonetheless, the activation of multiple PP2A complexes bodes well for the therapeutic efficacy of the sphingosine analogs.
While PP2A activation has been proposed as a therapeutic option for a range of cancers with inactivated PP2A, clinical trials have yet to eventuate. The demonstration that a nonphosphorylatable sphingosine analog, OSU-2S, is effective at eradiating AML stem cells while sparing normal immune cells is highly encouraging for clinical translation, and human trials are eagerly awaited.
Conflict-of-interest disclosure: The author declares no competing financial interests.


