Abstract
Since the publication of the Revised European-American Classification of Lymphoid Neoplasms in 1994, subsequent updates of the classification of lymphoid neoplasms have been generated through iterative international efforts to achieve broad consensus among hematopathologists, geneticists, molecular scientists, and clinicians. Significant progress has recently been made in the characterization of malignancies of the immune system, with many new insights provided by genomic studies. They have led to this proposal. We have followed the same process that was successfully used for the third and fourth editions of the World Health Organization Classification of Hematologic Neoplasms. The definition, recommended studies, and criteria for the diagnosis of many entities have been extensively refined. Some categories considered provisional have now been upgraded to definite entities. Terminology for some diseases has been revised to adapt nomenclature to the current knowledge of their biology, but these modifications have been restricted to well-justified situations. Major findings from recent genomic studies have impacted the conceptual framework and diagnostic criteria for many disease entities. These changes will have an impact on optimal clinical management. The conclusions of this work are summarized in this report as the proposed International Consensus Classification of mature lymphoid, histiocytic, and dendritic cell tumors.
Introduction
The publication of the Revised European-American Classification of Lymphoid Neoplasms (REAL) in 19941 and its subsequent validation across the world in 19972 represented a change of paradigm in the classification of these tumors. This classification provided a novel framework for the recognition of individual disease entities based on a constellation of features, including morphology, immune phenotype, clinical presentation, and genomics. This effort led to the World Health Organization (WHO) classification3 published in 2001, which extended the same conceptual approach to all hematopoietic and lymphoid neoplasms. The process was a joint effort of the Society for Hematopathology (SH) and the European Association for Haematopathology (EAHP) together with hematologists, oncologists, and scientists through joint Clinical Advisory Committees (CACs) at which collegial discussions led to broad consensus.4,5 The classification rapidly became the international standard, with publication of subsequent updates in 2008 and 2017.4-7 Since 2017, we have seen significant progress in the characterization of malignancies of the immune system, with many new insights provided by genomic studies. Initial planning and discussion for the current International Consensus Classification (ICC) took place in April 2021 at the twentieth meeting of the EAHP/SH. An international committee undertook the organization of the next CAC, which was held in September 2021. The subsequent discussions included 14 working groups (supplemental Table 1, available on the Blood Web site) with broad international participation. The conclusions of that meeting are summarized in this report with the proposal of the ICC (Table 1).
The definition of most entities remains unchanged, but criteria for diagnosis and recommended ancillary studies have been extensively refined. Some categories considered provisional in 2017 have now been upgraded to definite entities. Terminology for some diseases has been revised to adapt nomenclature to the current knowledge of their biology, but these modifications have been restricted to well-justified situations. Some categories such as multiple myeloma (MM) and Epstein-Barr virus (EBV)–positive T-cell lymphoproliferative disorders (LPDs) in children have undergone major revision. Major findings from recent genomic studies have had an impact on the conceptual framework of some diseases. This article will review the major revisions in the criteria and definition of mature lymphoid, histiocytic, and dendritic cell tumors (Tables 2–4).
Mature B-cell neoplasms
Chronic lymphocytic leukemia
The diagnostic criteria for chronic lymphocytic leukemia (CLL) and monoclonal B-cell lymphocytosis (MBL) are well established.5,8 Immunophenotype is determined by flow cytometry with a panel of CD19, CD5, CD23, and CD20 kappa and lambda that may be expanded in selected patients with CD43, CD79b, CD81, CD200, CD10, and ROR1 to clarify the diagnosis.8 The mutational status of the IGHV and TP53/17p alterations need to be evaluated at the time when patients require treatment.8 Although the (epi)genomic profile of CLL has been intensively investigated in the last decade,9-11 the clinical translation of the vast majority of the findings still requires further study. Factors likely to have significant clinical relevance include subclonal TP53 mutations with low variant allelic frequency (<10%), BCR stereotypes (eg, stereotypes 2 and 8), specific mutated genes (eg, NOTCH1, SF3B1, and BIRC3), and the IGLV3-21R110 mutation.12-17 Complex karyotype, defined as ≥3 aberrations, is currently applied in alignment with thresholds derived from other disease settings.8 However, in CLL, a distinct threshold of ≥5 abnormalities may better stratify very-high-risk patients.18 Although the prognostic impact of all these and other parameters has been shown in retrospective studies, clinical implementation will require methodologic evaluation, standardization, and validation in prospective studies.
Pathologists also recognize a tissue-based MBL, usually as an incidental nodal finding of an infiltrate of CLL-type cells without proliferation centers in individuals without significant lymphadenopathy.19,20 These patients usually have MBL in peripheral blood. At the other end of the CLL spectrum, the CAC emphasized the need to distinguish accelerated CLL from diffuse large B-cell (Richter) transformation, the latter containing sheets of large cells and not just expanded proliferation centers.21 The recent identification of reversible proliferations of sheets of large cells (Richter-like) in patients in which ibrutinib has been temporarily interrupted is a challenging situation to be considered in the interpretation of disease in such patients.22,23 These patients should be managed with caution and reevaluated after the reintroduction of ibrutinib.
The criteria for the diagnosis of B-cell prolymphocytic leukemia were also reviewed, and the group considered that the entity needs to be recognized only after rigorous exclusion of other lymphoid neoplasms, particularly transformation from CLL, mantle cell lymphoma (MCL), or splenic marginal zone lymphoma (SMZL).
Splenic marginal zone lymphoma
SMZL cannot be diagnosed on the basis of the extent of bone marrow or peripheral blood involvement alone. The presence of a clonal B-cell population in these locations with a phenotype consistent with MZL requires clinical or imaging evidence of splenic involvement for the diagnosis of an overt lymphoma. Distinction of SMZL from splenic diffuse red pulp small B-cell lymphoma requires evaluation of splenic histology. Next-generation sequencing (NGS) studies have identified recurrent mutations, including KLF2, NOTCH2, TNFAIP3, KMT2D, and TP53 among others.24-26 Sequencing studies may support the diagnosis of SMZL, but the overlap with other entities makes NGS profiles inadequate for establishing a diagnosis in isolation. Recent data have described genetically defined subsets of SMZL with prognostic differences.27MYD88 mutations remain valuable in the differential diagnosis of SMZL vs lymphoplasmacytic lymphoma (LPL).
Lymphoplasmacytic lymphoma and immunoglobulin M monoclonal gammopathy of undetermined significance
The diagnostic criteria for LPL have been refined from the revised fourth edition of the WHO classification.7 In keeping with the diagnostic criteria proposed by the International Workshop on Waldenström’s Macroglobulinemia, a diagnosis of LPL may be rendered in patients with abnormal lymphoplasmacytic aggregates in the bone marrow and evidence of clonal B cells and plasma cells, even when the aggregates represent <10% of cellularity of the trephine biopsy.28 Molecular studies for MYD88 and CXCR4 mutations are strongly encouraged in the workup of suspected LPL. MYD88 mutations in the Toll-interleukin-1R resistance (TIR) domain are found in >90% of LPLs; the L265P variant is predominantly present, although non-L265P variants may rarely be present. Although MYD88 mutations are not specific, they help with the diagnosis of LPLs in an appropriate clinicopathologic context.29-31 A small percentage of patients with LPL have MYD88 wild-type with alternative mutations downstream of MYD88 in the NFKB signaling pathway.32,33 Absence of an MYD88 mutation therefore does not completely exclude the diagnosis of LPL. CXCR4 mutations are identified in up to 40% of patients with LPL, particularly LPL with nonsense variants, which have been associated with symptomatic hyperviscosity and resistance to ibrutinib therapy.34-36 However, this effect is complex and requires further research as treatment options expand.
The diagnosis of immunoglobulin M monoclonal gammopathy of undetermined significance (IgM MGUS) is established in patients who have IgM paraprotein with <10% bone marrow plasma cells and who lack lymphoplasmacytic B-cell aggregates sufficient for a diagnosis of LPL.29,37 Two subtypes of IgM MGUS are now further distinguished32: IgM MGUS of plasma cell type and IgM MGUS, not otherwise specified (NOS). The rare IgM MGUS of plasma cell type is considered a precursor of MM and is defined as showing clonal plasma cells without a detectable B-cell component and with wild-type MYD88. This category also includes patients with t(11;14)(q13;q32) or other cytogenetic abnormalities typical of MM. The remaining patients with IgM MGUS, NOS include all those with an MYD88 mutation, those with detectable monotypic or monoclonal B cells but without abnormal lymphoplasmacytic aggregates diagnostic of LPL, and those who lack evidence of other small B-cell neoplasms. Routine fluorescence in situ hybridization (FISH) studies and MYD88 mutation analysis are recommended to identify the rare tumors more likely to progress to MM rather than LPL or other B-cell neoplasms.
Primary cold agglutinin disease is recognized as a new diagnostic category, distinct from LPL or IgM MGUS. This disease lacks the MYD88 L265P mutation but displays recurrent trisomies of chromosomes 3, 12, and 18 and recurrent mutations in KMT2D and CARD11.38-40
Plasma cell neoplasms
Clinicians participating in the CAC strongly supported the term “multiple myeloma” over “plasma cell myeloma.” MM is a genetically heterogeneous disease with 2 main groups defined by cytogenetics. Specifically, 40% to 50% of patients show recurrent IGH translocations with a variety of partner genes, whereas up to 55% of patients with MM lack IGH translocations and are characterized by hyperdiploidy, with a small subset of patients not falling into either category.41,42 These primary genetic abnormalities are present in precursor conditions and persist throughout the disease course. They are associated with prognosis, treatment response, and other clinical and phenotypic features and have a strong correlation with the gene expression profile (GEP).41,43-45 Therefore, MM can be formally divided into mutually exclusive diagnostic groups: (1) MM, NOS and (2) MM with recurrent genetic abnormalities, including MM with CCND family translocations, MM with MAF family translocation, MM with NSD2 translocation, and MM with hyperdiploidy. Detection of t(4;14), t(14;16), and secondary changes, including del(17p), amp1q, and del(1p) identifies patients with high-risk disease.46-48 Currently, interphase FISH is the technique of choice for cytogenetic characterization, and consensus FISH panels for MM have been published.47 The role of mutational analysis requires further study, particularly given the frequent subclonal evolution and spatial heterogeneity in MM.45,49-51
MGUS of the non-IgM type is a virtually universal precursor to MM.52 Although most patients with MGUS are asymptomatic, several conditions associated with clonal Ig secretion in the absence of overt malignancy have been recognized and have been termed “monoclonal gammopathy of renal significance (MGRS) or monoclonal gammopathy of clinical significance (MGCS).”53,54 However, these do not represent separate disease entities; instead, they are descriptive terms that can be added as a clinical feature to the underlying diagnosis (eg, MGUS).
Smoldering or asymptomatic MM, defined as lacking features of active MM (SLiM CRAB criteria: SLiM: 60% or more clonal plasma cells, light chains, and magnetic resonance imaging; CRAB: increased calcium level, renal dysfunction, anemia, and destructive bone lesions) or amyloid light chain (AL) amyloidosis,37 exhibits broad variability in progression to active MM. Risk stratification with models proposed for this situation should be used to select patients suited for early therapeutic intervention.55
Solitary plasmacytomas of bone and primary extramedullary plasmacytomas are plasma cell neoplasms with low to moderate risk for progression to MM.56,57 Because minimal marrow involvement detected by flow cytometry (ie, clonal plasma cells present but <10%) is of major prognostic importance, particularly with solitary plasmacytomas of bone, this feature should be incorporated into the diagnosis of these entities.56,58
For clarity, primary amyloidosis should be termed “Ig light chain (AL) amyloidosis” and needs to be separated from localized AL amyloidosis (also termed “amyloid tumor”), a rare disorder with excellent prognosis and rare progression to systemic AL amyloidosis.59-61
Marginal zone lymphomas
There is no indication for separately classifying extranodal MZLs of mucosa-associated lymphoid tissue (MALT lymphoma) based on site of presentation except for cutaneous MZL, which is now designated separately as a lymphoproliferative disorder (see “Cutaneous lymphomas” below). The clinical management approach, however, may differ between anatomic sites (eg, gastric MALT). In nodal MZL, significant heterogeneity is recognized, but there is no consensus on further alterations to the diagnostic criteria. The diagnosis of large-cell transformation of MZL should continue to rest on the finding of diffuse sheets of large cells.
Follicular lymphoma
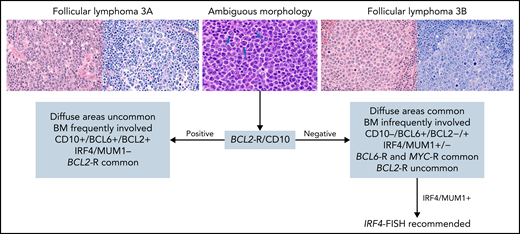
For follicular lymphoma (FL), the consensus was to retain morphologic grading (grades 1-2, 3A, and 3B) according to previously described criteria.7 Whether patients with grade 3A have a more adverse prognosis and deserve different management than those with grades 1 to 2 remains debatable62-64 and needs to be re-evaluated, given evolving non-cytotoxic therapeutic approaches. Grade 3B clearly differs in its clinical behavior, and patients are usually managed similarly to those with diffuse large B-cell lymphoma (DLBCL).65,66 Hence, distinction between grade 3A and 3B is critical, and some higher-grade lesions are difficult to classify.67 The consensus was that the presence of BCL2 rearranged (BCL2-R) and CD10 positivity (detectable by FISH) both favor FL grade 3A (Figure 1). In addition, patients with grade 3B-expressing IRF4/MUM1 should be evaluated for IRF4 alterations,68,69 especially in younger patients. Routine screening for MYC-R is not recommended for detecting the rare patients with FL who carry both BCL2-R and MYC-R, although those patients might have a more aggressive outcome.70-73 Proliferation index using Ki-67 staining can be specified, but it has uncertain clinical significance in isolation74 and is not required for grading. Routine molecular testing is currently unnecessary, but it can be useful in selected patients for differential diagnosis (eg, pediatric-type FL, plasmacytic differentiation, MZL, BCL2-R–negative patients). Detection of EZH2 mutations provides additional information when treatment with an EZH2 inhibitor is being considered.75 Use of an NGS panel for clinical prognostication such as the m7-FLIPI (mutation status of 7 genes [EZH2, ARID1A, MEF2B, EP300, FOXO1, CREBBP, and CARD11] along with the FL International Prognostic Index)76 improves risk stratification but remains investigational.
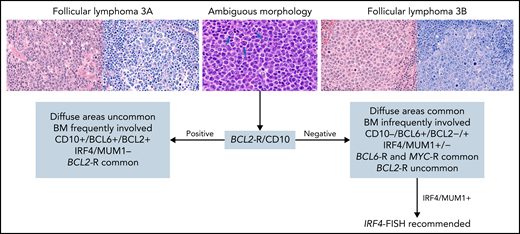
Suggested diagnostic studies in FL grade 3. Upper left: Cells from FL grade 3A are shown with hematoxylin and eosin (H&E) and Giemsa stains. Note the admixture of centrocytes and centroblasts (>15 per high power field) highlighted in the Giemsa stain. Upper right: Cells from FL grade 3B are shown with H&E and Giemsa stains. The follicles are composed of sheets of centroblasts with open chromatin, several nucleoli, and abundant basophilic cytoplasm highlighted with the Giemsa stain. Upper middle: Cells from FL with ambiguous morphology are shown. They are medium-size with open chromatin but inconspicuous nucleoli unlike centroblasts (arrows) and without the cytologic features of centrocytes. With ambiguous morphology (blue arrow), the presence of BCL2 rearrangement and/or CD10 expression favors the diagnosis of FL grade 3A; if both are absent, a diagnosis of FL grade 3B is favored. In patients who have FL grade 3B with IRF4/MUM1 expression, IRF4-FISH analysis is recommended to exclude the diagnosis of large B-cell lymphoma with IRF4 rearrangement. Original magnification ×400. BM, bone marrow.
Suggested diagnostic studies in FL grade 3. Upper left: Cells from FL grade 3A are shown with hematoxylin and eosin (H&E) and Giemsa stains. Note the admixture of centrocytes and centroblasts (>15 per high power field) highlighted in the Giemsa stain. Upper right: Cells from FL grade 3B are shown with H&E and Giemsa stains. The follicles are composed of sheets of centroblasts with open chromatin, several nucleoli, and abundant basophilic cytoplasm highlighted with the Giemsa stain. Upper middle: Cells from FL with ambiguous morphology are shown. They are medium-size with open chromatin but inconspicuous nucleoli unlike centroblasts (arrows) and without the cytologic features of centrocytes. With ambiguous morphology (blue arrow), the presence of BCL2 rearrangement and/or CD10 expression favors the diagnosis of FL grade 3A; if both are absent, a diagnosis of FL grade 3B is favored. In patients who have FL grade 3B with IRF4/MUM1 expression, IRF4-FISH analysis is recommended to exclude the diagnosis of large B-cell lymphoma with IRF4 rearrangement. Original magnification ×400. BM, bone marrow.
Nodal FL negative for BCL2-R is heterogeneous, both genetically and clinically.77-79 The specific subtype of BCL2-R–negative, CD23+ follicle center lymphoma was proposed as a provisional new entity based on correlation of CD23 with STAT6 mutation, low-stage disease, and often a predominant diffuse growth pattern. This variant typically presents with localized inguinal involvement.
Pediatric-type FL remains a clearly defined entity with recurrent genomic alterations and excellent prognosis with conservative management.80-83 Distinguishing pediatric-type FL from FL grade 3B remains critical. Recent work has suggested that pediatric-type FL may be related to the pediatric variant of MZL, which had been listed as provisional in the classification.84 Testicular FL, recognized as a new distinct entity of FL in young boys, shares pathological and clinical features with pediatric-type FL, because most patients can be managed conservatively, without systemic chemotherapy.85,86
Large B-cell lymphoma with IRF4 rearrangement, upgraded now to a definite entity, is most common in children and young adults and usually has at least a partially follicular growth pattern.69 However, the same disease is not commonly seen in adults. FISH for IRF4-R must be performed for diagnosis. Patients lacking demonstrable rearrangements should have evidence of either IGH or IGK/IGL breaks. Detection of IRF4 mutation may support the diagnosis.69IRF4-R can occur in other aggressive B-cell lymphomas associated with BCL2-R or MYC-R, mainly in adults, and in this context, it is not specific for the entity.69
Mantle cell lymphoma
The CCND1 translocation with IG genes is the genetic hallmark of MCL. Some patients with the same morphology, phenotype, and SOX11 expression as that found in conventional MCL lack CCND1 rearrangements but have (sometimes cryptic) CCND2 or CCND3 translocations.87-90 These patients must also be diagnosed as having MCL. CCND2 and CCND3 translocations by FISH or messenger RNA overexpression should be demonstrated in these patients, because immunohistochemistry for these cyclins is not discriminant.91 The presence of t(11;14)(q13;q32) may also be a secondary event in the progression of some mature B-cell lymphomas. Patients with that abnormality should not be diagnosed as having MCL.92-97CCND1 rearrangement has also been found in large B-cell lymphomas associated with MYC and BCL2 or BCL6 translocations. The negativity of CD5 and SOX11 and the presence of mutations uncommon in MCL favor the diagnosis of DLBCL over MCL.96 Conversely, MYC may be rearranged in bona fide MCL, usually with blastoid or pleomorphic morphology and aggressive behavior.98-101 Using the term “double-hit” (DH) MCL for these patients is not recommended and those patients should not be included in the high-grade B-cell lymphoma (HGBCL) category. Some of these patients may be SOX11 negative or express terminal deoxynucleotide transferase (TdT).100 Genomic studies may help in the differential diagnosis with other lymphomas.
MCLs with more aggressive or indolent behavior need to be identified. The unfavorable outcome of blastoid or pleomorphic variants, high Ki-67 (≥30%), and TP53 deletions or mutations have been extensively confirmed and should be evaluated, preferably at diagnosis, in all patients.102-106 Determination of the Ki-67 proliferative index is currently based on visual inspection according to previously described criteria.105 Whether the evaluation of proliferation or other quantitative parameters suggested in this ICC proposal will benefit from quantitative flow cytometry, RNA technologies, or computer-assisted image analysis in clinical practice will require standardization and validation studies. Genomic complexity is also associated with worse outcome, but further studies are needed before incorporation into clinical practice.99,107,108 At the other end of the spectrum, most leukemic non-nodal MCLs (nnMCLs) are clinically indolent, although the acquisition of TP53 alterations and genomic complexity confer an adverse prognosis. MCL in these patients is considered a subtype of MCL because t(11;14) is acquired in precursor B cells as in conventional MCL.99,107,108 Recognition of nnMCL relies on a combination of clinical and pathological characteristics. Features that favor this diagnosis are non-nodal or limited nodal (≤3 cm) presentation, negative or low SOX11 expression (<10%), CD23 and CD200 positivity, and hypermutated IGHV (<98%).108-112 Absence of ATM mutations or deletions and CCND1 mutations are also features of nnMCL.99 MCL with isolated gastrointestinal involvement usually has an indolent behavior and should be clinically recognized, although more data are needed to determine significance.113-115
Diffuse large B-cell lymphomas
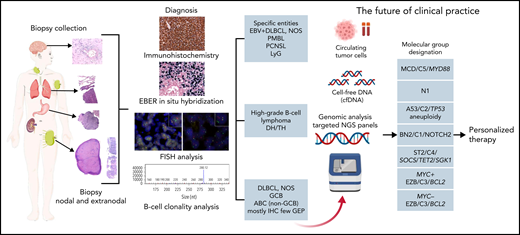
DLBCL, NOS encompasses all patients with nodal and extranodal large B-cell lymphoma that do not belong to a specific diagnostic category (Table 1). It is not a single disease but a collection of morphologically, genetically, and clinically different diseases. Therefore, it can be subdivided into morphologic variants, phenotypic variants, and molecular or genetic categories. The role of morphologic variants (centroblastic, immunoblastic, and anaplastic) and phenotypic variants (DLBCL, CD5+,116-119 and DLBCL double expressor [MYC/BCL2])120-122 should be deemphasized. These variants have (weak) adverse prognostic impact and do not reflect true biological subgroups but rather represent the end results of different biological pathways. The conference considered that at this time, the cell-of-origin designation in DLBCL, NOS123,124 should be maintained. The cell-of-origin distinction is a basic biological division of DLBCL with prognostic impact that can be widely deployed using either IHC (germinal center B-cell–like [GCB] and non-GCB patients) or gene expression (GCB, activated B-cell–like [ABC], and unclassified patients) algorithms. However, the largely disappointing results of trials of first-line treatment of DLBCL, NOS that incorporated targeted agents and use cell-of-origin for patient selection underscore the lack of sufficient detail for this binary classification and highlight the importance of a more molecularly based approach.125-130 Recently, molecular and cytogenetic profiling studies have independently identified 5 to 7 new functional genetic subgroups of DLBCL, which strongly emphasizes the validity of this concept but fails to classify all patients (Figure 2).131-134 A combination of cell-of-origin and molecular subclassification may provide more precise patient stratification for developing future clinical trials.135 Overall, cell-of-origin is retained for the present time with the expectation that transition to a molecular genetic classification will be feasible in the near future.
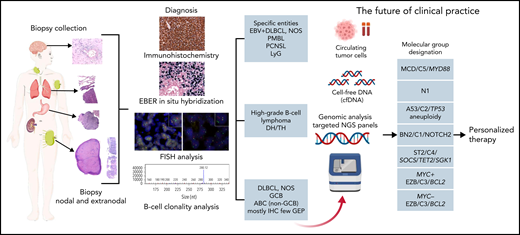
Algorithm for the diagnostic workup of aggressive B-cell lymphomas. The current algorithm for diagnosing aggressive large B-cell lymphomas starts with a biopsy collection from a lymph node (excision or needle biopsy) or a biopsy of an extranodal site. The diagnosis of the different lymphoma entities is based on a combination of morphology, immunophenotype, EBER in situ hybridization, FISH analysis, and B-cell clonality analysis. Advances in the understanding of DLBCL herald a transition to a molecular genetic classification (red arrow). This genetic classification is based on mutational profile, somatic copy number alterations, and structural variants. The depicted molecular subtypes were identified in 3 different studies indicating that these subgroups reflect true biological differences.131,132,134 On the basis of these molecular studies, a predictor model was developed that dissects the cell-of-origin and stratifies further the molecular classification into 7 genetic subtypes with apparently clinical relevance.133 The acronyms indicate the names given in the different studies to the same identified biological group.
Algorithm for the diagnostic workup of aggressive B-cell lymphomas. The current algorithm for diagnosing aggressive large B-cell lymphomas starts with a biopsy collection from a lymph node (excision or needle biopsy) or a biopsy of an extranodal site. The diagnosis of the different lymphoma entities is based on a combination of morphology, immunophenotype, EBER in situ hybridization, FISH analysis, and B-cell clonality analysis. Advances in the understanding of DLBCL herald a transition to a molecular genetic classification (red arrow). This genetic classification is based on mutational profile, somatic copy number alterations, and structural variants. The depicted molecular subtypes were identified in 3 different studies indicating that these subgroups reflect true biological differences.131,132,134 On the basis of these molecular studies, a predictor model was developed that dissects the cell-of-origin and stratifies further the molecular classification into 7 genetic subtypes with apparently clinical relevance.133 The acronyms indicate the names given in the different studies to the same identified biological group.
An intensely debated but ultimately unresolved issue is whether an umbrella term such as “extranodal lymphoma ABC (non-GCB) type” should be created for (some) extranodal DLBCLs. This would primarily (but not exclusively) include patients with DLBCL that arises in immune-privileged sites such as primary central nervous system lymphoma (PCNSL) and primary DLBCL of the testis but possibly also primary cutaneous DLBCL, leg type, primary breast type, intravascular large B-cell lymphoma, and primary adrenal lymphomas. The rationale is that most of the lymphomas in these locations are non-GCB/non-ABC type, share biology, and seem to display common molecular features such as the high prevalence of MYD88L265P and CD79B mutations that characterize the DLBCL MCD/C5 genetic subgroup (Figure 2).135-140 In particular, PCNSL and primary DLBCL of the testis share both clinical and molecular features, and for this reason, primary DLBCL of the testis is now considered a distinct entity (Tables 1 and 2). Although grouping the extranodal lymphomas arising in immune-privileged sites certainly is a reasonable proposal, there are also many caveats, including the fact that particularly in some anatomic sites, these lymphomas are heterogeneous, and in many settings, the pathologist may have incomplete data regarding the presence of other sites of disease. In the end, although many participants were inclined to group several of the extranodal DLBCL entities and variants, the majority felt that such a subcategorization of DLBCL is premature, and recognition of specific entities will be better captured by upcoming molecular categorization integrated with more traditional criteria.
Provisional subtypes of large B-cell lymphoma
The 2016 WHO classification recognized the provisional entity, Burkitt-like lymphoma (BLL) with 11q aberration, identified originally as a lesion clinically and pathologically resembling Burkitt lymphoma (BL) but lacking MYC-R. The patients are more frequently children and young adults with a good prognosis. Subsequent studies have demonstrated the morphology and phenotype of these tumors to be more variable than originally described, including patients with mainly centroblastic-type large cells.141-143 Importantly, genetic studies also suggest the disease is distinct from BL and is closer to conventional DLBCL with GCB derivation harboring more complex karyotypes and the absence of typical BL mutations.141-145 This provisional entity has now been renamed “large B-cell lymphoma with 11q aberration” (Figure 3). Chromosome 11q gains and losses typical of patients with this abnormality can be identified by using FISH strategies. Although some studies suggest that only 11q loss may be acceptable, more information is needed before a strong recommendation can be made. Chromosomal microarray is required if FISH is equivocal for the typical pattern of gains and losses.141
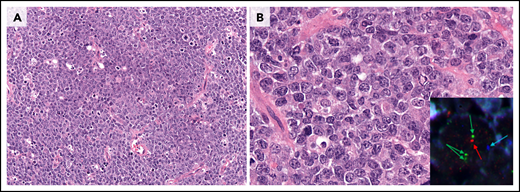
Large B-cell lymphoma with 11q aberration. (A) Low power view of large-cell morphology, abundant mitoses, and the characteristic starry-sky pattern with abundant macrophages with coarse apoptotic bodies (original magnification ×200; H&E stain). (B) Higher magnification reveals the large centroblastic morphology of the tumor cells (original magnification ×400; H&E stain). Inset: FISH analysis demonstrated the typical 11q alterations (blue, centromere; red, 11q24 loss; green, 11q23 gain; ×1000). The cytology of the cells might be medium-size to large-size cells. The morphology and mutational profile justify the change in the name of this entity (previously, Burkitt-like lymphoma with 11q aberration).
Large B-cell lymphoma with 11q aberration. (A) Low power view of large-cell morphology, abundant mitoses, and the characteristic starry-sky pattern with abundant macrophages with coarse apoptotic bodies (original magnification ×200; H&E stain). (B) Higher magnification reveals the large centroblastic morphology of the tumor cells (original magnification ×400; H&E stain). Inset: FISH analysis demonstrated the typical 11q alterations (blue, centromere; red, 11q24 loss; green, 11q23 gain; ×1000). The cytology of the cells might be medium-size to large-size cells. The morphology and mutational profile justify the change in the name of this entity (previously, Burkitt-like lymphoma with 11q aberration).
“HHV-8 and EBV-negative primary effusion-based lymphoma” is a new provisional entity recognized on the basis of unifying features that include presentation in elderly HIV-negative patients with medical conditions that lead to fluid overload, which suggests chronic serosal stimulation in pathogenesis. About 60% of the patients have been reported in Japan, and they often have a history of hepatitis C infection.146-148 These patients usually have a good prognosis with reported spontaneous regression or cure with drainage alone. Most tumors exibit centroblastic or immunoblastic morphology and express at least 1 B-cell marker. Other HHV-8–negative effusion-based lymphomas occur and are biologically and clinically heterogeneous. These should be classified as one of the well-defined lymphomas presenting as an effusion.
Large B-cell lymphoproliferative disorders and viral agents
EBV-positive polymorphic B-cell LPD, NOS is a term used for EBV-positive B-cell proliferations with or without known immunodeficiency that cannot be more precisely categorized. The term should be reserved for patients with altered lymph node architecture and a polymorphic infiltrate that do not fulfill criteria for the diagnosis of lymphoma or there is uncertainty because of a small size or low-quality biopsy.149,150 EBV-positive B-cell proliferations should be classified as lymphoma if the criteria of a well-defined EBV-associated lymphoma are fulfilled (eg, EBV-positive DLBCL, NOS, and plasmablastic lymphoma). In tissues with low to modest numbers of EBV-positive B cells without distortion of the nodal architecture, the term “EBV reactivation” is preferred. EBNA2 immunostaining is recommended in this or other clinical settings because it supports EBV latency pattern III, which suggests an underlying immunodeficiency. It is negative in most EBV-positive tumors in otherwise healthy people.
EBV-positive DLBCL, NOS, is an aggressive lymphoma that can present over a wide age range; however, patients younger than age 45 years have a better prognosis.151-153 By definition, >80% of the malignant cells should express EBER.152,154,155 The morphology is variable. A T-cell/histiocyte-rich large B-cell lymphoma-like pattern is frequently seen in younger patients and is associated with a better prognosis. In adults, the pattern may be monomorphic or polymorphic, but these patterns do not have prognostic impact.152,154-156 The differential diagnosis with EBV-positive classic Hodgkin lymphoma (CHL) can be challenging; however, expression of B-cell markers in >50% of the tumor cells, extranodal presentation, and/or EBV latency III favors the diagnosis of EBV-positive DLBCL, NOS. Extended B-cell antibody panels are critical in this setting.157 DLBCL associated with chronic inflammation and fibrin-associated DLBCL remain discrete entities, separate from EBV-positive DLBCL, NOS.
“EBV-positive mucocutaneous ulcer” was introduced in the 2016 WHO classification as a provisional entity,5 but it is now considered a definite entity.149,156,158-160 These are solitary lesions, usually in the oropharyngeal mucosa. Cutaneous and gastrointestinal presentations are usually associated with iatrogenic immunosuppression. In patients with ≥2 skin lesions, the term “EBV-positive B-cell polymorphic LPD,” or when appropriate, “EBV-positive DLBCL, NOS,” or other specific type of EBV-positive lymphoma or LPD is preferred.160,161
Lymphomatoid granulomatosis (LyG) is a rare angiocentric and angiodestructive LPD composed of a variable number of EBV-positive B cells admixed with numerous reactive T cells. Pulmonary involvement is required for the diagnosis.162 Although the disease is well defined, there are significant overlapping features with other immunodeficiency-related EBV-positive B-cell LPDs.162,163 Isolated central nervous system (CNS) or gastrointestinal tract involvement by an EBV-positive lesion resembling LyG is observed usually in the context of known causes of defective immune surveillance (EBV latency III).164,165 In this scenario, the diagnosis of EBV-positive polymorphic B-cell LPD or EBV-positive DLBCL, NOS should be rendered.
HHV-8–associated lymphoproliferations include multicentric Castleman disease, HHV-8 germinotropic LPD, HHV-8–positive DLBCL, NOS, primary effusion lymphoma (PEL), and extracavitary PEL.166 There are significant overlapping features among these disorders.166,167 PEL and extracavitary PEL in HIV-positive patients are usually HHV-8 positive and EBV positive; however, in elderly HIV-negative individuals, EBV is usually negative.166,168-170 In extracavitary presentations, the diagnosis of HHV-8–positive DLBCL, NOS should be favored in EBV-negative patients with cytoplasmic IgM lambda and/or associated with multicentric Castleman disease.171
High-grade B-cell lymphomas
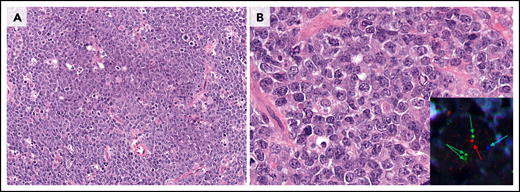
The 2016 WHO classification included 2 categories of HGBCL: HGBCL, NOS, and HGBCL with MYC and BCL2 and/or BCL6 rearrangements (DH or triple-hit [TH]).5 HGBCL-DH now comprises 2 groups: HGBCL with MYC and BCL2 rearrangements (with or without BCL6 rearrangement) (HGBCL-DH-BCL2) and a new provisional entity, HBGBL with MYC and BCL6 rearrangements (HGBCL-DH-BCL6). HGBCL-DH-BCL2 and HGBCL-DH-BCL6 entities continue to exclude FL, and the morphology (large-cell or high-grade cytology) should be reported (Figure 4).

Morphologic characterization of highly proliferative B-cell lymphomas. (A-B) This DLBCL, NOS has many mitotic figures, but many of the neoplastic cells are typical large transformed cells that do not resemble either BL cells or B lymphoblasts. Chromosomal analysis showed a complex karyotype, but there was no evidence of MYC or BCL2 rearrangement. (C-D) This HGBCL, NOS is composed of relatively small blastoid-appearing cells with many mitotic figures, reminiscent of a B-lymphoblastic leukemia/lymphoma. TdT was negative. It had a complex karyotype that included t(14;18)(q32;q21) and i(17)(q10). (E-F) This HGBCL with MYC and BCL6 rearrangements (without evidence of IGH::BCL2) resembles BL with intermediate-size transformed cells and a starry-sky appearance with scattered tingible body macrophages. The cytospin (inset) demonstrated cytoplasmic vacuoles. Unlike classic BL, it was BCL2 protein positive and had only equivocal CD10 positivity. All panels were stained with H&E except for the inset stained with Wright-Giemsa stain. Original magnification ×400 for panels A, C, and E; original magnification ×1000 for panels B, D, and F and inset.
Morphologic characterization of highly proliferative B-cell lymphomas. (A-B) This DLBCL, NOS has many mitotic figures, but many of the neoplastic cells are typical large transformed cells that do not resemble either BL cells or B lymphoblasts. Chromosomal analysis showed a complex karyotype, but there was no evidence of MYC or BCL2 rearrangement. (C-D) This HGBCL, NOS is composed of relatively small blastoid-appearing cells with many mitotic figures, reminiscent of a B-lymphoblastic leukemia/lymphoma. TdT was negative. It had a complex karyotype that included t(14;18)(q32;q21) and i(17)(q10). (E-F) This HGBCL with MYC and BCL6 rearrangements (without evidence of IGH::BCL2) resembles BL with intermediate-size transformed cells and a starry-sky appearance with scattered tingible body macrophages. The cytospin (inset) demonstrated cytoplasmic vacuoles. Unlike classic BL, it was BCL2 protein positive and had only equivocal CD10 positivity. All panels were stained with H&E except for the inset stained with Wright-Giemsa stain. Original magnification ×400 for panels A, C, and E; original magnification ×1000 for panels B, D, and F and inset.
Studies performed since the 2016 WHO classification support HGBCL-DH-BCL2 as an aggressive lymphoma of GCB origin with distinct biology from other GCB-DLBCL, NOS and HGBCL-DH-BCL6.172-177 It can occur in patients with or without previous FL. Data to support distinct biology in patients with HGBCL-DH-BCL6 are less compelling172,173; however, it has been retained as a provisional entity to allow for continued study based on the poor outcomes seen in some studies.175,178-181 Although pseudo-DH lymphomas (MYC-R with BCL6 partner) account for up to 30% of patients with HGBCL-DH-BCL6,182 strategies to identify this are not essential at this time. Neither copy number increase nor amplification of these genes is sufficient to substitute for rearrangement in these categories.183-186 Furthermore, the significance of the MYC partner gene remains controversial; MYC-R with both IG and non-IG partners is included at present.180,187,188
Although HGBCL, NOS is acknowledged as a heterogeneous category, it remains in this classification as a diagnosis of exclusion for tumors which are not HGBCL-DH but which have intermediate-size cells, often with blastoid or Burkitt-like cytology (Figure 3) but cannot be classified as DLBCL or BL.189,190 These patients are rare, and the diagnosis can be made only on well-fixed and preserved specimens because large-cell cytology must be excluded. DLBCL with starry-sky morphology and/or a high proliferation index does not merit recategorization as HGBCL, NOS.
Previously, TdT expression in HGBCL or DLBCL was sufficient to reclassify the disease in these patients as lymphoblastic leukemia/lymphoma.7 However, the mutational landscape of TdT-positive HGBCL now supports the inclusion of this disease as a mature lymphoma with “expression of TdT” noted in the diagnostic line.191-193 Distinction between patients with this disease and those with acute leukemia requires thorough phenotypic and genetic evaluation.191-194
Diagnostic criteria for BL remain largely unchanged. However, data have emerged to segregate TdT-positive patients from those with BL. These rare patients have an immature B-cell phenotype and molecular features of precursor B cells, including evidence of IG::MYC translocation arising from aberrant variability, diversity, and joining (VDJ) recombination, frequent lack of a productive IGH rearrangement, DNA methylation patterns similar to those in other pre–B-cell acute leukemias, and recurrent NRAS and KRAS mutations.195 On the basis of these data, designating these patients as having B-lymphoblastic leukemia/lymphoma with MYC-R is appropriate to recognize their biology and allow clinicians to consider appropriate treatment options (see Arber et al in this series).196,197
Hodgkin lymphomas
The CAC conference discussed key issues related to the classification of Hodgkin lymphomas and patients with borderline diagnostic criteria. The conference concluded that new terminology is warranted for nodular lymphocyte-predominant Hodgkin lymphoma (NLPHL), based on major biological and clinical differences with CHL and with close relationship to T-cell/histiocyte-rich large B-cell lymphoma.198 The term “nodular lymphocyte predominant B-cell lymphoma” (NLPBL) was accepted by consensus. The value of identifying variant histology in NLPBL was recognized, with the suggestion that typical patients with Fan patterns A, B, and C or grade 1 be distinguished from Fan patterns D, E, and F or grade 2.199 Patients falling within grade 2 generally show loss of a well-formed nodular pattern and increased infiltration by T cells with a reduction of background small B cells. Patients with grade 2 histology may warrant treatment for DLBCL, but clinical features should play a role in treatment decisions.200 Rare examples of NLPBL are EBV-positive with uncertain clinical implications.201
The major subtypes of CHL remain unchanged. A standard immunohistochemical panel using CD30, CD15, IRF4/MUM1, PAX5, CD20, CD3, and LMP1 or EBER in situ hybridization is advised. Additional immunohistochemical or clonality studies may be warranted in the setting of atypical histological or clinical features.
A major topic of discussion related to the criteria for mediastinal gray zone lymphoma (MGZL). This term is preferred over what was previously designated “B-cell lymphoma, unclassifiable,” with features intermediate between DLBCL and CHL. A diagnosis of MGZL requires both morphologic (high density of tumor cells) and immunophenotypic criteria (at least 2 B-cell markers with strong expression).202,203 Patients with otherwise typical nodular sclerosis CHL with variable expression of CD20 are still designated as having CHL, although a close biological relationship to primary mediastinal large B-cell lymphoma remains.204 Sequential primary mediastinal large B-cell lymphoma and nodular sclerosis CHL reinforce the concept of MGZL, because these diseases have been demonstrated to be of common clonal origin. However, clinical and genomic data indicate that most patients with non-mediastinal GZL are distinct from those with MGZL, and they should be diagnosed as having DLBCL, NOS. Finally, nearly all patients with EBV-positive DLBCL, while they may harbor admixed Hodgkin/Reed-Sternberg-like cells, differ at the genomic level from patients with MGZL and should be retained within the category of EBV-positive DLBCL.152,205
Mature T-cell and NK-cell neoplasms
At the CAC meeting, discussion of the T-cell and NK-cell neoplasms focused on those areas in which new insights into the pathogenesis and clinical behavior have occurred. Thus, only a subset of this large and diverse group of tumors will be covered.
EBV-related mature T-cell and NK-cell neoplasms
EBV-positive T-cell and NK-cell LPDs in children are now separated into 4 major groups: hydroa vacciniforme (HV) LPD, severe mosquito bite allergy, chronic active EBV (CAEBV) disease, and systemic EBV-positive T-cell lymphoma of childhood (Table 4). All occur with increased frequency in Asia and Latin America. HV LPD presents with skin lesions on sun-exposed areas with EBV-infected T or NK cells and very high levels of EBV DNA in blood.7,206,207 This disease was previously referred to as hydroa vacciniforme-like LPD; however, it is now known that all HV lesions have EBV. Some patients, especially white patients, have stable disease involving only the skin (classic HV LPD)208 whereas others, especially Asians209 and Hispanics have concomitant systemic EBV-positive T cells or NK cells involving internal organs (systemic HV LPD).206,210,211 This latter group eventually requires treatment similar to that for CAEBV disease.212 CAEBV disease is a progressive disorder that lasts 3 or more months during which patients have markedly increased levels of EBV DNA in the blood and infiltration of organs by EBV-infected lymphocytes in the absence of a known immunodeficiency.213-215 This illness was previously referred to as CAEBV infection; however, because most adults are chronically infected with EBV, the term “CAEBV disease” is preferred. Previously, CAEBV disease included EBV-infected T, NK, or B cells. Many patients with B-cell CAEBV have been diagnosed with underlying primary immunodeficiency; therefore, CAEBV should include only T- or NK-cell disease.216 Some patients in South America present with facial edema, high levels of EBV DNA in T or NK cells in the blood, and EBV in internal organs; these patients should be classified as having CAEBV disease and not HV LPD.217 New genetic studies have shown that CAEBV disease shares somatic mutations (eg, DDX3X and KMT2D) similar to those in T- and NK-cell lymphomas, indicating that it is a premalignant condition. Furthermore, the EBV genome harbors intragenic deletions common in various EBV-associated neoplastic disorders but not detected in reactive conditions such as infectious mononucleosis, which suggests an important role of these mutations in EBV-associated neoplasia.218
“Primary nodal EBV-positive T- or NK-cell lymphoma” is a rare disease introduced in the 2017 WHO classification as a variant of peripheral T-cell lymphoma (PTCL), NOS.7 New findings have led to the designation of this lymphoma as a provisional entity.219 It presents more commonly in elderly and/or immunodeficient patients, lacks nasal involvement, and is more often of T-cell rather than NK-cell lineage.220,221 This lymphoma is characterized by a dismal outcome, low genomic instability, upregulation of immune pathways (checkpoint protein programmed death-ligand 1 [PD-L1]) that promote immune evasion, and downregulation of EBV micro RNAs.222,223
Extranodal T-cell and NK-cell neoplasms involving the gastrointestinal tract
The two main types of primary intestinal T-cell lymphomas are enteropathy-associated T-cell lymphoma (EATL), which may be preceded by refractory celiac disease, and monomorphic epitheliotropic intestinal T-cell lymphoma (MEITL).7 Novel immunophenotypic and genomic data reinforce their distinction.224 Expression of SYK is absent in EATL.225 Most patients with EATL are T-cell receptor (TCR)–silent, whereas most patients with MEITL express the TCR and derive more frequently from gamma-delta T cells than from alpha-beta T cells.225-229 MEITL has highly recurrent alterations in SETD2, resulting in defective trimethylation of H3K36 and frequent mutations in STAT5B, JAK3, TP53, and GNAI2.228-232 Type II refractory celiac disease is a precursor of EATL and has therefore been added to the classification. EATL and type II refractory celiac disease have frequent gain-of-function mutations in STAT3 and JAK1.229,233-235 Intestinal T-cell lymphoma, NOS remains an entity for overtly malignant primary intestinal EBV-negative T-cell lymphomas, after EATL, MEITL, and other PTCL entities, notably adult T-cell lymphoma/leukemia, have been excluded.
Two groups of indolent LPDs of the gastrointestinal tract are recognized, according to their T-cell or NK-cell derivation.236-239 The clonal nature of the T-cell disease (indolent clonal T-cell LPD of the gastrointestinal tract), which variably express CD4 and/or CD8, is further supported by the finding of gene alterations in a subset of the patients.240-242 The intestinal NK-cell proliferation formerly referred to as NK-cell enteropathy237 or lymphomatoid gastropathy,243 is now recognized as a neoplasm designated as indolent NK-cell LPD of the gastrointestinal tract.236 These 2 entities are EBV negative and have a limited propensity to infiltrate the gastrointestinal tract with a superficial distribution.
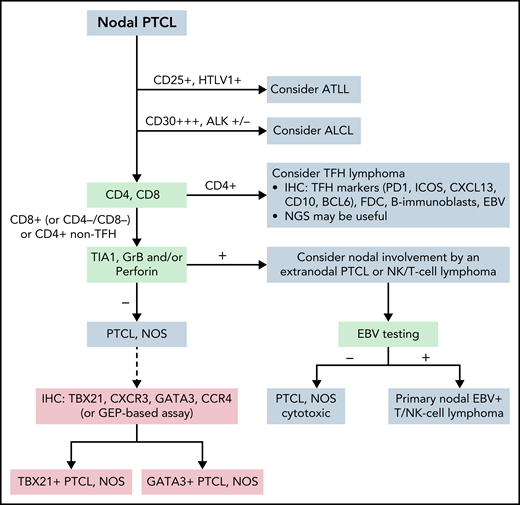
Peripheral T-cell lymphoma, NOS
PTCL, NOS is mainly a nodal lymphoma that remains a diagnosis of exclusion (Figure 5). Two molecular subgroups—PTCL-TBX21 and PTCL-GATA3—have been identified based on their GEP resembling T helper type 1 (Th1) and Th2 cells, respectively. The PTCL-GATA3 subgroup has been associated with a worse outcome in some studies and has greater genomic complexity.244 The PTCL-TBX21 subgroup has better prognosis, fewer copy number alterations, and more frequent mutations in genes that regulate DNA methylation.244 These subgroups may be recognized by using an immunohistochemistry-based algorithm with 4 markers (TBX21, CXCR3, GATA3, and CCR4).245-247 In addition, the expression of cytotoxic molecules delineates a subgroup of aggressive PTCLs, NOS which tend to occur in patients with impaired immunity and mostly cluster to PTCL-TBX21.244,248 Designation of PTCL, NOS according to the molecular subgroups is not routinely incorporated into clinical diagnosis and requires further studies for clinical validation.
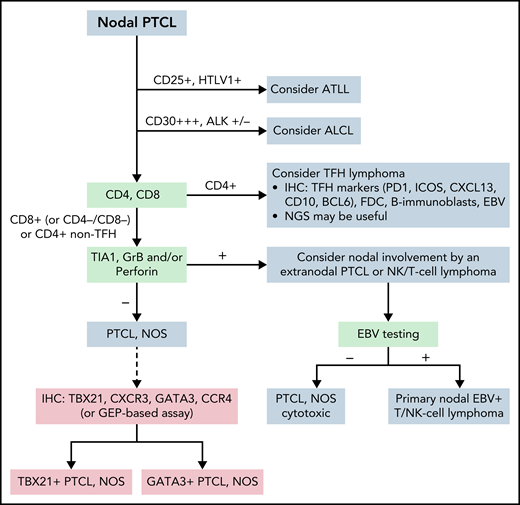
Algorithm for the classification workup of nodal PTCLs. The current algorithm for diagnosing PTCL requires immunophenotypic study with a panel of markers that, together with viral analysis (HTLV1, EBV), will orient the pathologist to consider and diagnose specific entities. In ambiguous cases, sequencing studies may help diagnose some entities, particularly follicular helper T-cell lymphoma. PTCL, NOS is established when other specific entities are excluded. Phenotypic analysis or analysis by GEP may subdivide patients with PTCL, NOS, but this subclassification is not routinely incorporated into clinical diagnosis and requires further studies for clinical validation. ATLL, adult T-cell leukemia/lymphoma; GrB, granzyme B; Per, perforine.
Algorithm for the classification workup of nodal PTCLs. The current algorithm for diagnosing PTCL requires immunophenotypic study with a panel of markers that, together with viral analysis (HTLV1, EBV), will orient the pathologist to consider and diagnose specific entities. In ambiguous cases, sequencing studies may help diagnose some entities, particularly follicular helper T-cell lymphoma. PTCL, NOS is established when other specific entities are excluded. Phenotypic analysis or analysis by GEP may subdivide patients with PTCL, NOS, but this subclassification is not routinely incorporated into clinical diagnosis and requires further studies for clinical validation. ATLL, adult T-cell leukemia/lymphoma; GrB, granzyme B; Per, perforine.
Follicular helper T-cell lymphoma
Since the discovery that T follicular helper (TFH) cells represent the normal cell counterpart of the neoplastic cells in angioimmunoblastic T-cell lymphoma (AITL),249,250 a larger subset of nodal PTCLs not diagnostic of AITL have been found to express markers of normal TFH cells and/or are more GEP-enriched than normal TFH cells.251 The 2016 WHO classification created an umbrella category of “nodal lymphomas of TFH origin,” covering 3 entities, namely AITL, follicular helper T-cell lymphoma, and PTCL with TFH phenotype showing a diffuse or T-zone pattern without follicular dendritic cell (FDC) expansion.5 A TFH phenotype was defined by the expression of 2 or preferably 3 phenotypic markers of normal TFH cells, among which those most widely used are CD10, BCL6, CXCL13, PD1, and ICOS.251-253 Multiple studies have reinforced the notion that these 3 entities are unified by a common genetic landscape in addition to a TFH immunophenotype.251,252,254 Loss-of-function mutations in genes that regulate DNA and histone methylation, specifically TET2 (present in about 80%), and/or DNMT3A (present in 30%-40%) of the patients, and several lines of evidence, indicate that AITL in many instances develops on a background of clonal hematopoiesis. Other alterations include a highly recurrent RHOAG17V hotspot mutation, mutations in IDH2R172, and mutations in genes involved in TCR signaling.251,255IDH2 mutations seem to be restricted to AITL with characteristic large clear-cell cytomorphology.253 Several pathogenic fusions involving CD28, ICOS, and VAV1 have been reported.256 Overall, the combinatory pattern of mutations in genes related to epigenetics and TCR signaling is a feature common to all nodal lymphomas of TFH origin. These lymphomas show a better response to histone deacetylase inhibitors compared with other PTCLs, which suggests the clinical relevance of the TFH phenotype.257-259 For these reasons, the ICC unifies systemic lymphomas of TFH origin as a single entity—TFH lymphoma—with 3 subtypes: angioimmunoblastic-type (AITL), follicular-type, and NOS. By definition, this entity is restricted to patients with primary nodal or systemic disease and excludes primary cutaneous small or medium CD4+ T-cell LPDs or other specified subtypes of cutaneous lymphomas with a TFH phenotype.260 The criteria for distinguishing the 3 TFH lymphoma subtypes remain essentially unchanged and rely mainly on morphology and immunoarchitecture, especially the tumor microenvironment and distribution of FDCs. For establishing the TFH immunophenotype, which is critical for the diagnosis of TFH lymphomas of follicular type and NOS, we recommend the use of a 5-marker panel. Because RHOAG17V or IDH2R172 are so characteristic of TFH lymphomas, especially of the AITL type, NGS studies are valuable in supporting a diagnosis of TFH lymphoma.261
Anaplastic large-cell lymphoma
ALK-negative anaplastic large-cell lymphoma (ALCL) remains a distinct systemic entity. Primary cutaneous ALCL and breast implant–associated ALCL must be excluded from this category. Criteria for the diagnosis remain unchanged. The disease should resemble ALK-positive ALCL with a common pattern, have strong uniform CD30 expression, and lack ALK expression. DUSP22-R ALK-negative ALCL is now defined as a genetic subtype of systemic ALK-negative ALCL based on distinct morphologic, phenotypic, genomic, and epigenetic features.192,262-266DUSP22-R is present in 19% to 30% of ALK-negative ALCLs, and FISH testing is recommended in all ALK-negative ALCLs. DUSP22-R ALCL tends to have a favorable prognosis, but in some patients, it may behave aggressively, probably related to high International Prognostic Index (IPI) score and other high-risk clinical features.262,265-267TP63 rearrangements are associated with poor prognosis. Patients with the rare co-existing TP63-R and DUSP22-R require further study.268 Patients with JAK2-R may have a disease that resembles CHL, which presents a potential diagnostic challenge.269
Breast implant–associated ALCL was upgraded to a definite entity based on its unique clinical, genomic, and molecular features distinct from other ALCLs.270-275 Pathologic and clinical staging is important to determine prognosis and assess the need for chemotherapy. Formation of a mass lesion, capsular invasion, and lymph node involvement are adverse prognostic features.276,277 Comprehensive capsulectomy sampling,278 margin evaluation, and use of tumor-node-metastasis (TNM) staging criteria (T1: in situ, tumor cells in seroma and/or on capsular luminal surface; T2: early capsule infiltration; T3: aggregates or sheets infiltrating capsule; T4: infiltration beyond capsule) are recommended.277
Cutaneous lymphomas
Several significant changes are being introduced in the ICC regarding primary cutaneous lymphomas. Primary cutaneous marginal zone lymphoproliferations will now be recognized as distinct from other MALT lymphomas. They will now be called “primary cutaneous marginal zone LPD” rather than “lymphoma” because of their extremely indolent behavior; disease-specific survivals approach 100% without requiring aggressive therapies. However, cutaneous recurrences are common. Primary cutaneous marginal zone LPDs show significant differences compared with MALT lymphomas at other sites.7,279-285 Two subtypes of this disorder are recognized, largely but not exclusively identified on the basis of whether they are heavy chain, class-switched, or IgM positive.7,283,285-287 Approximately three-quarters of primary cutaneous marginal zone LPDs are class-switched and predominantly IgG+ with up to ∼40% expressing IgG4.285,288 These patients often have other unique features, including abundant reactive T cells and peripherally located plasma cells. Caution must be taken with IgM+ tumors to exclude non-cutaneous primary disease.280,283,287 Rare patients with class-switched disease are similar to those with IgM+ primary cutaneous marginal zone LPDs and have features more like those in patients with typical MALT lymphomas.287 Molecular and genetic studies of both primary cutaneous marginal zone and primary cutaneous follicle center lymphomas have further supported their recognition as distinct entities and have potential diagnostic utility.289-291
Primary cutaneous DLBCL, leg type remains a distinct entity. Many patients share the molecular and cytogenetic features seen in DLBCL of MCD/C5 type, a finding also shared with PCNSL, primary DLBCL of the testis, and intravascular large B-cell lymphoma.133,135,138,292,293 About 25% of the latter are restricted to the skin and reported to have a better prognosis than the systemic variant.138,294,295 Primary cutaneous DLBCL, leg type, is considered to be of the non-GCB/ABC type, but one study reported that these patients may be more heterogeneous in terms of their cell-of-origin with frequent MYD88 and CD79B mutations.296 However, this study includes a large number of unclassified patients by GEP and triple-positive patients by Hans algorithm (CD10, BCL6, and IRF4/MUM1). Consistent with the recognition that some high-grade B-cell lymphomas can be TdT positive, some patients have been reported with TdT positivity, which should not prompt reclassification of their disease as a B-cell lymphoblastic neoplasm.297,298
There are new molecular and cytogenetic data regarding a variety of cutaneous T-cell lymphomas of biologic and, to some extent, clinical and potential therapeutic interest. This includes specific findings such as the germline HAVCR2 mutations in many patients with subcutaneous panniculitis-like T-cell lymphomas299-301 and also the more extensive genetic and epigenetic findings in other cutaneous T-cell lymphomas, including mycosis fungoides and Sézary syndrome.302 However, there is only one significant change in the classification of the primary cutaneous T-cell lymphomas. Consistent with a general trend to greater conservatism, primary cutaneous acral CD8+ T-cell lymphoma, in spite of its very monotonous and atypical morphologic appearance, is now classified as a primary cutaneous acral CD8+ T-cell LPD, largely because of its very indolent course and general need for only local type therapies or even just observation.303-305 Although ∼20% of patients do have a local or more extensive recurrence, only 1 patient with extracutaneous spread is described, and a 100% survival rate is reported independent of treatment modality.281,305 Some do still advise clinical caution.306 Aiding in their distinction from other CD8+ cutaneous T-cell lymphomas is their characteristic dot-like CD68 positivity in the neoplastic cells.307 A rare CD4+CD8+ patient has been reported.308
Immunodeficiency-associated lymphoproliferative disorders
The iatrogenic immunodeficiency-associated LPDs include posttransplant LPD (PTLD), and the separately designated LPD arising in patients receiving methotrexate or other immunosuppressive agents.7 Although there are some common histologic features shared by EBV-positive B-cell LPDs in diverse clinical settings,150 the consensus was to retain PTLD as a separate subgroup based in part on major differences in clinical management. Subclassification of PTLDs, not all of which are EBV- positive, remains unaltered from the 2017 WHO classification.7 Although studies of other iatrogenic immunodeficiency-associated LPDs are much more limited, it is recommended that they be classified in a fashion analogous to PTLD. This was a topic not discussed in great detail at the CAC and requires further study.
Histiocytic and dendritic cell neoplasms
The classification of histiocytic and dendritic cell neoplasms has matured in recent years.309 Delineation of B-cell and T-cell lymphomas developed from a concerted effort to relate the tumors to developmental and functional subsets of the normal immune system,310 while many of the histiocytoses were initially thought to be reactive or inflammatory conditions. The list includes Erdheim-Chester disease, Rosai-Dorfman-Destombes disease, and Langerhans cell histiocytosis.
Study of the molecular pathogenesis of these neoplasms indicates convergence along a common pathway, with frequent mutations in the mitogen-activated protein kinase (MAPK) pathway.311,312 A smaller subset of patients shows evidence of activation of the PI3K signaling pathway.313 These insights have led to advances in therapy, with the introduction of targeted therapy through inhibition of RAS, RAF, MEK, and MTOR.309,313 Nevertheless, many of the observed mutations are not specific to any individual entity. For example, BRAFV600E mutations can be encountered in all members of the disease family, including isolated Langerhans cell histiocytosis, systemic Erdheim-Chester disease, and histiocytic and dendritic cell sarcomas. ALK-positive histiocytosis is a relatively new addition to the list of histiocytic neoplasms,314,315 and involves rearrangements of ALK, leading to activation of signaling pathways. First described by Chan et al,316 the cells have a mature histiocytic phenotype and often have foamy cytoplasm. Patients who present in infancy usually have systemic disease, whereas patients who present as adults usually have more localized disease.
EBV-positive inflammatory FDC/fibroblastic reticular cell (FRC) tumor is an indolent proliferation of stromal cells of mesenchymal origin not derived from hematopoietic stem cells. Neoplastic cells are EBV positive and are associated with a rich inflammatory background. Spleen and liver are the most common sites, but the tumors also arise in other extranodal locations.317-319
Conclusion
The clinicopathological, molecular, and genomic information generated on lymphoid neoplasms in the last 5 years provides solid grounds for refining the diagnostic criteria of several entities, consolidating the status of categories previously defined as provisional, and identifying some new entities. The explosion of genomic data is having an impact on our understanding of these diseases and is starting to be introduced into routine clinical practice for diagnosis and management strategies. However, in many areas, incorporation of these data into general practice requires further validation and standardization.
Acknowledgments
We dedicate this report to the memory of Paul Kleihues (21 May 1936–17 March 2022), former director of the International Agency for Research on Cancer (IARC) (1994-2003), a visionary leader who created the modern WHO Blue Book series for the classification of tumors.
E.S.J., L.M.S., and J.I.C. were supported by the intramural research programs of the Center for Cancer Research, National Cancer Institute (E.S.J., ZIA SC 000550; L.M.S., ZIABC011780) and the National Institute of Allergy and Infectious Diseases (J.I.C., ZIAAI000058). Funding for the work of the CAC was received from many sources as previously cited.5
Authorship
Contribution: E.C., E.S.J., J.R.C., L.Q.-M., S.H.S., K.C.A., P.B., L.C., L.d.L., S.D., A. Dogan, A.L.F., F.F., J.W.F., P. Gaulard, P. Ghia, S.M.H., R.L.K., G.S., J.S.-M., J.F.S., S.P.T., J.M.V., E.Z., R.A., S.A., W.-Y.A., C.B., L.B., W.C.C., J.I.C., F.d’A., A. Davies, B.F., I.M.G., J.G., J.G.G., E.D.H., B.S.K., W.-S.K., S.K., A.S.L., C.L., G.L., J.P.L., M.P.L., A.L.-G., M.V.M., E.M., A.M.M., F.M., S.N., M.N., A.P., S.A.P., M.P., B.P., V.R., S.T.R., B.S., L.S., M.A.S., S.M.S., L.M.S., C.T., T.T., W.H.W., T.Y., P.-L.Z., M.D., D.W.S., J.N.W., and A.D.Z. contributed to the contents of this manuscript; and E.C., E.S.J., J.R.C., M.D., L.Q.-M., D.W.S., S.H.S., J.N.W., and A.D.Z. coordinated the work of the Clinical Advisory Committee and edited the final manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Elias Campo, Hematopathology Unit, Laboratory of Pathology, Hospital Clinic of Barcelona, Villarroel 170, 08036 Barcelona, Spain; e-mail: ecampo@clinic.cat; and Elaine S. Jaffe, Hematopathology Unit, Laboratory of Pathology, 10 Center Dr, Room 3S 235, National Cancer Institute, National Institutes of Health, Bethesda, MD 20892; e-mail: ejaffe@mail.nih.gov.
The online version of this article contains a data supplement.