Abstract
Tissue factor pathway inhibitor (TFPI) is a Kunitz-type protease inhibitor with three tandem inhibitory domains (K1, K2, and K3) that regulates the initial reactions of the extrinsic blood coagulation pathway through K1 and K2. In the present study, the effect of thrombin on TFPI in a purified system was first examined using recombinant TFPI from Chinese hamster ovary (CHO) cells. TFPI was inactivated by thrombin with cleavage of three peptide bonds, Lys 254-Thr 255 in the C-terminal basic region, Arg 107-Gly 108 (reactive site toward factor Xa in K2), and Lys 86-Thr 87 between K1 and K2. Then, degradation of radiolabeled TFPI by thrombin was examined in two systems: (1) mixed with plasma and then tissue factor (TF ) and calcium ion, and (2) mixed with fibrinogen and then thrombin. TFPI degradation was detected in serum from normal plasma and more extensively from antithrombin (AT)-depleted plasma by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Significant radioactivity was found in the clot after coagulation of the plasma, which decreased after 20 hours' incubation. These changes were more prominent in AT-depleted plasma than in normal plasma. When TFPI lacking the C-terminal basic region was used instead of full-length TFPI, most of the radioactivity was found in serum rather than in fibrin clots. Incorporation of TFPI into the fibrin clot was prevented by a synthetic C-terminal peptide of TFPI. Similar results were obtained after mixing radiolabeled TFPI with fibrinogen and then thrombin in the presence of calcium ion or EDTA. These results demonstrate a novel degradation pathway of TFPI, ie, incorporation into fibrin via the C-terminal basic region and degradation by thrombin (possibly fibrin-bound thrombin).
TISSUE FACTOR pathway inhibitor (TFPI) is a Kunitz-type protease inhibitor with three tandem inhibitory domains (K1, K2, and K3), a negatively charged N-terminus, and a positively charged C-terminus.1 TFPI inhibits the initial reactions of the tissue factor (TF )-mediated blood coagulation pathway, interacting with factor Xa via K2 and with the TF/factor VIIa complex via K1.1 TFPI is synthesized mainly by endothelial cells and exists on the endothelium and also in plasma in a free form and lipoprotein-associated forms. Whereas many studies of TFPI have been made using recombinant TFPI from Escherichia coli, yeast, and Chinese hamster ovary (CHO) cells, our knowledge of plasma TFPI is limited. The molecular size of plasma TFPI was reported to be heterogeneous; 34 kD in low-density lipoprotein and 41 kD in high-density lipoprotein, the latter of which was a mixed disulfide complex between TFPI and apolipoprotein A-II.2 Recombinant TFPI (rTFPI) produced by mammalian cells and TFPI released by heparin in vivo were approximately 43 kD.3-5 Broze et al6 reported that there are carboxy-terminal–truncated forms of TFPI in human plasma, lacking the C-terminal basic region and even a portion of K3. These truncated forms of TFPI showed reduced inhibitory activity. It has been demonstrated that leukocyte elastase preferentially cleaves a peptide bond between K1 and K2,7 8 but the proteases responsible for truncation of plasma TFPI have not yet been reported.
In the present study, we examined the effect of thrombin on rTFPI from CHO cells, in which we determined the amino acid sequence and carbohydrate structure.9 We found that thrombin significantly inactivated the inhibitory activity of rTFPI, cleaving three peptide bonds. Thrombin is a serine protease that plays a central role in thrombosis and hemostasis. It converts fibrinogen to fibrin, and activates blood coagulation factors V, VIII, and XIII and protein C in the presence of thrombomodulin.10 11 Thrombin can also aggregate platelets and modulate the function of the various cells.
To examine the significance of TFPI inactivation by thrombin, we mixed radiolabeled rTFPI with plasma and then TF, or with fibrinogen and then thrombin. We found that radiolabeled rTFPI was initially associated with fibrin clots and then released by degradation with thrombin after prolonged incubation. We also found that significant amounts of rTFPI in plasma were incorporated into the fibrin clot when coagulation was induced by the addition of TF. In the present report, we propose a novel degradation pathway of TFPI, namely incorporation of TFPI into fibrin clots and degradation by fibrin-bound thrombin, and discuss the in vivo significance of the degradation pathway.
MATERIALS AND METHODS
Human plasma.Human normal plasma was obtained from healthy donors and stored at −80°C. Human antithrombin (AT)-depleted plasma was prepared using an anti-AT antibody column as described previously.12 AT activity was not detected from this preparation. Human plasminogen-depleted plasma and plasminogen- and AT-depleted plasma were prepared using Lysine-Sepharose (Pharmacia Biotechnology, Uppsala, Sweden) from normal plasma or AT-depleted plasma according to the manufacturer's instructions. Human factor XIII–deficient plasma was obtained from George King Bio-Medical Inc (Overland Park, KS).
Electrophoresis.Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) was performed using Phast Gel Gradient 8-25, Phast Gel High Density (both from Pharmacia Biotechnology) or a Multi Gel containing 10% to 20% polyacrylamide (Daiichi Pure Chemicals, Tokyo, Japan) according to the Laemmli method.13 Molecular weight standards were obtained from Pharmacia Biotechnology. Protein bands were visualized by staining with Coomassie brilliant blue. The gels were scanned by a GT-9000 scanner (Epson, Nagano, Japan), and the data were analyzed using Version 2.0 of the image-analyzing program, Mac Bas (Fuji Film, Tokyo, Japan). The intensity of each band was calculated relative to the intensity of bands from the 8-hour thrombin digest of TFPI as described later herein.
Effect of thrombin on TFPI.Human rTFPI was prepared from CHO cells as described previously3 and incubated with human thrombin (Protogen AG, Läufelfingen, Switzerland) in 50 mmol/L Tris HCl, pH 7.5, containing 0.15 mol/L NaCl (TBS) at 37°C in the presence or absence of 1 mmol/L D-Phe-Pro-Arg-chloromethylketone (Calbiochem, La Jolla, CA), 5 μmol/L hirudin (American Diagnostica Inc, Greenwich, CT), or 75 μmol/L argatroban (Mitsubishi Chemical, Tokyo, Japan). Final concentrations of TFPI and thrombin were 2.6 and 0.05 mg/mL, respectively. At the specified times, aliquots were removed from the reaction mixture and stored immediately at −80°C. Subsamples were then analyzed by SDS-PAGE or assayed for residual TFPI inhibitory activity toward factor Xa and factor VIIa/TF as described later. The effect of thrombin on rTFPI was also examined in the presence of 5 mmol/L calcium, 10 U/mL heparin (Sigma, St Louis, MO), or 0.1 mg/mL soluble thrombomodulin (kindly supplied by Dr Koji Suzuki, Department of Molecular Pathobiology, Mie University School of Medicine, Mie, Japan).
Effect of TFPI on thrombin activity.Thrombin inhibition by TFPI was measured using Boc-Val-Pro-Arg-4-methylcoumarin-7-amide ([Boc-Val-Pro-Arg-MCA] Peptide Institute Inc, Minoh, Osaka, Japan) as a substrate. Fifteen microliters of rTFPI (0 to 31.7 μmol/L) was incubated with 15 μL thrombin (5 nmol/L) at 37°C. After 1 hour, sample solution (20 μL) was added to a cuvette thermostated at 37°C. Into this solution, 10 μL 20-mmol/L substrate was injected, and the reaction was initiated. Final concentrations were 50 pmol/L for thrombin, 0 to 317 nmol/L for rTFPI, and 0.2 mmol/L for fluorogenic substrate in TBS containing 0.1% bovine serum albumin. Thrombin activity was expressed as the initial increase in fluorescence intensity of 7-amino-4-methylcoumarin (AMC) at 380 nm (excited at 440 nm).
Effect of plasmin on TFPI.TFPI was incubated with human plasmin in 50 mmol/L Tris HCl, pH 7.5, containing 0.15 mol/L NaCl (TBS) at 37°C. Plasmin was prepared using Lysine-Sepharose from normal plasma according to the manufacturer's instruction. Final concentrations of TFPI and plasmin were 2.6 and 0.05 mg/mL respectively. Samples were then analyzed by SDS-PAGE.
Effect of thrombin or plasmin on Boc-Val-Leu-Lys-MCA.Twenty microliters of 10-mmol/L fluorogenic substrate was added to 960 μL TBS containing 0.1% BSA in a cuvette thermostated at 37°C. Into this solution, 20 μL thrombin or plasmin was injected, and the reaction was initiated. Final concentrations for thrombin and plasmin were 0 to 22 nmol/L. The final concentration for the fluorogenic substrate was 0.2 mmol/L. Enzyme activity was expressed as the initial increase in fluorescence intensity of AMC at 380 nm (excited at 440 nm).
TFPI inhibition of factor VIIa/TF.Factor VIIa/TF inhibition by rTFPI was measured by a two-stage method based on the capacity of TFPI to inhibit factor VIIa/TF activity toward factor X activation, as previously described.14 Briefly, in the first stage, the sample to be tested was incubated with a solution containing human factor VIIa (kindly supplied by Dr Tomohiro Nakagaki, Chemo-Sero-Therapeutic Research Institute, Kumamoto, Japan), rabbit brain thromboplastin for PT reagent (Kokusai Shiyaku, Kobe, Japan), and calcium. After 7 minutes' incubation, this mixture was mixed with human factor X (kindly supplied by Dr Tomohiro Nakagaki, Chemo-Sero-Therapeutic Research Institute). After 15 minutes' incubation at 37°C, a peptidyl fluorogenic substrate (Z-Pyr-Gly-Arg-MCA) for factor Xa (Peptide Institute Inc) was added as a second step. The increase in fluorescence intensity of AMC was monitored by measuring fluorescence intensity with excitation at 380 nm and emission at 440 nm using a centrifugal autoanalyzer (COBAS FARA II; Roche, Basel, Switzerland).
TFPI inhibition of factor Xa.Inhibition of factor Xa by TFPI was measured using Z-Pyr-Gly-Arg-MCA as a substrate. Each sample of rTFPI (final concentration, 282 nmol/L) was incubated with 5 nmol/L human factor Xa (Enzyme Research Laboratories, South Bend, IN) in TBS containing 0.1% bovine serum albumin in a cuvette thermostated at 37°C. The reaction was initiated by addition of 10 μL 20-mmol/L substrate. Final concentrations were 2.8 nmol/L for rTFPI, 3.0 nmol/L for factor Xa, and 0.2 mmol/L for fluorogenic substrate. Release of AMC was monitored by measuring fluorescence intensity with excitation at 380 nm and emission at 440 nm. The data were collected at 60 points/min using a fluorescence spectrometer (model F4500; Hitachi, Tokyo, Japan).
Reverse-phase high-performance liquid chromatography.Reverse-phase high-performance liquid chromatography (HPLC) was performed on a Cosmosil 5C18-300 column (4.6 × 100 mm; Nacalai Tesque) coupled with Waters instrumentation. After samples were injected, the column was washed with a solution of 0.05% trifluoroacetic acid for 10 minutes. TFPI fragments were eluted with a linear concentration gradient formed by the solutions containing 36% and 54% acetonitrile with a flow rate of 0.5 mL/min.
Amino acid sequence analysis.Amino acid sequences of rTFPI fragments were determined using a protein sequencer (model 476A; Applied Biosystems, Foster City, CA). Amino acid sequence data analysis was performed using a data analysis program (610A data analysis system; Applied Biosystems). For sequence analysis of proteins in SDS-PAGE gels, each band was electroblotted onto a problot membrane (Applied Biosystems). The corresponding band was cut from the membrane and subjected to sequence analysis.
Effect of TF-induced coagulation on rTFPI in plasma.rTFPI or rTFPI-C (rTFPI lacking the C-terminal basic region, Rockford, IL) was radiolabeled with 125I using Iodo-gen (Pierce Chemical) according to the manufacturer's instructions. Normal or AT-depleted plasma, plasminogen-depleted plasma, and both plasminogen and AT-depleted plasma (100 μL) were mixed with 10 μL 125I-rTFPI (10 μg/mL) and preincubated at 37°C. One hundred microliters of TF (Thromborel S; Hoechst Japan, Tokyo) containing calcium ion with or without argatroban (Mitsubishi Chemical) was added to this mixture, and the reaction was started. The final concentration of 125I-TFPI was 900 ng/mL. After allowing the mixture to stand for 15 minutes or 20 hours, samples were centrifuged to separate the serum from the fibrin clot. Serum was mixed with an equal volume of 10% 2-mercaptoethanol and 5% SDS, and heated at 95°C for 15 minutes. After the fibrin clot was washed with TBS, 100 μL 5% 2-mercaptoethanol and 2.5% SDS were added. After heating at 95°C, the clot was completely dissolved. Radioactivity in the serum and clot was measured using an automatic gamma counter (Cobra 5003; Packard Japan, Tokyo). The samples were then assayed on 10% to 20% SDS gels. Autoradiography of the gel was performed using an image plate and Bio Image Analyzer (Bas 2000; Fuji Film).
Effect of thrombin-induced fibrin formation on TFPI in fibrinogen solution.Two hundred microliters of fibrinogen (grade L; Kabi, Stockholm, Sweden) solution in TBS (3 mg/mL) was mixed with 10 μL 125I-rTFPI (18 μg/mL). Calcium chloride or EDTA was added to the mixture, with the final concentration 5 mmol/L calcium ion or 5 mmol/L EDTA. The reaction was started by adding 5 μL thrombin (2 μg/mL). After 15 minutes or 20 hours, fibrin was removed with a small wooden stick and rinsed with TBS. The fibrin clot was dissolved in 500 μL 5% 2-mercaptoethanol and 2.5% SDS. Radioactivity in the supernatant, in the washing buffer, and in the fibrin solution was measured.
Enzyme-linked immunosorbent assay of TFPI in plasma and serum.One hundred microliters of plasma was mixed with 100 μL TF containing calcium ion (Thromborel S). After allowing the mixture to stand for 15 minutes, samples were centrifuged to separate the serum from the fibrin clot. TFPI antigen in the original plasma and serum was measured by an ELISA kit specific for free-form TFPI (Free TFPI ELISA kit; Chemo-Sero-Therapeutic Research Institute).15
Effect of synthetic peptides on TFPI binding to fibrin.The C-terminal basic peptide of TFPI with the sequence from amino acid residues 242 to 266 of human TFPI with additional cysteine residue at the C-terminal was synthesized using a peptide synthesizer (Model 430A; Applied Biosystems). The Gly 212-Phe 243 peptide in the K3 domain was prepared as described previously.3 C-terminal cysteine residue of C-terminal peptide was carboxyaminomethylated16 and desalted using reverse-phase HPLC using a Cosmosil 5C18-300 column (Nacalai Tesque). The peptide concentration was determined by amino acid analysis using the Hitachi L-8500A amino acid analyzer. One hundred microliters of fibrinogen solution in TBS (6 mg/mL) was mixed with 100 μL of each peptide (0 to 300 μmol/L). Then, 10 μL 125I-rTFPI (18 μg/mL) was added to the mixture, and the reactions were started by adding 5 μL thrombin (2 μg/mL). After 15 minutes, fibrin was removed as already described and radioactivity in the supernatant, in the washing buffer, and in the fibrin solution was measured.
RESULTS
Degradation of TFPI by thrombin.Figure 1 shows SDS-PAGE results for rTFPI incubated with thrombin at an enzyme to inhibitor ratio of 1:50 (wt/wt). In the absence of reducing reagent, the rTFPI band decreased with the increase of two major bands of molecular weight 15 kD (band 2) and less than 15 kD (band 1) (Fig 1A). A faint band at 35 kD was also observed after 8 hours' incubation. The additional band with molecular weight 35 kD (band 3) newly appeared in the presence of reducing reagent (Fig 1B). After 8 hours' incubation, the rTFPI band almost disappeared in the presence of reducing reagent. This result suggests that thrombin cleaved the peptide bonds of the polypeptide chain bridged with disulfide bonds, in addition to the peptide bonds outside the disulfide bridges. To exclude the possibility that contaminated proteases in the preparation of thrombin cleaved TFPI, effects of thrombin inhibitors on cleavage were examined: 1 mmol/L D-Phe-Pro-Arg-chloromethylketone, 5 μmol/L hirudin, or 75 μmol/L argatroban completely inhibited cleavage of rTFPI (data not shown). Addition of calcium ion or thrombomodulin did not affect the degradation pattern of TFPI on SDS-PAGE, but when heparin was added to the reaction mixture, band 1 disappeared (data not shown). When the enzyme to inhibitor ratio was varied from 1:100 to 1:1, a similar degradation pattern of TFPI on SDS-PAGE was observed, although degradation of TFPI was promoted with the increase of enzyme concentration (data not shown). The amidolytic activity of thrombin was not affected by a 6,200-fold molar excess of rTFPI (data not shown).

SDS-PAGE analysis of rTFPI incubated with thrombin. rTFPI was incubated with thrombin in TBS at 37°C with the final concentrations of 2.6 mg/mL and 0.05 mg/mL, respectively. SDS-PAGE of the digest was performed as described in Materials and Methods. (A), Nonreducing condition; (B), reducing condition. See text for nomenclature of band 1, band 2, and band 3.
SDS-PAGE analysis of rTFPI incubated with thrombin. rTFPI was incubated with thrombin in TBS at 37°C with the final concentrations of 2.6 mg/mL and 0.05 mg/mL, respectively. SDS-PAGE of the digest was performed as described in Materials and Methods. (A), Nonreducing condition; (B), reducing condition. See text for nomenclature of band 1, band 2, and band 3.
Degradation of TFPI by thrombin was associated with a loss in the ability of rTFPI to inhibit the activities of factor Xa and factor VIIa/TF (Fig 2). Under the same conditions described in Fig 1, thrombin reduced the ability of rTFPI to inhibit both factor Xa and factor VIIa/TF with the increase in incubation time. About 50% of TFPI activity was lost after 4 hours' incubation. No loss of inhibitory activity was observed when rTFPI was incubated in the absence of thrombin (data not shown).

Effect of thrombin on the inhibitory activities of rTFPI toward factor Xa and TF/factor VIIa. The reaction mixture as described in Fig 1 was subjected to the analysis of TFPI inhibitory activity. The residual TFPI activity was expressed as the percent relative to that of intact rTFPI. (•), Inhibitory activity toward factor Xa; (○), inhibitory activity toward TF/factor VIIa.
Effect of thrombin on the inhibitory activities of rTFPI toward factor Xa and TF/factor VIIa. The reaction mixture as described in Fig 1 was subjected to the analysis of TFPI inhibitory activity. The residual TFPI activity was expressed as the percent relative to that of intact rTFPI. (•), Inhibitory activity toward factor Xa; (○), inhibitory activity toward TF/factor VIIa.
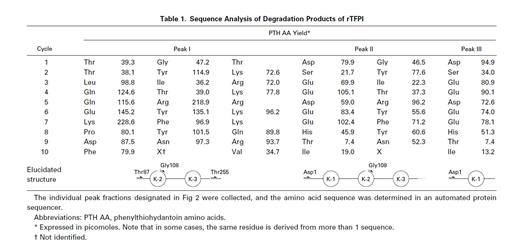
When the 9.5-hour digest of rTFPI with thrombin was subjected to reverse-phase HPLC, three distinct peaks were separated (Fig 3A). They were subjected to N-terminal amino acid sequence analysis (Table 1).
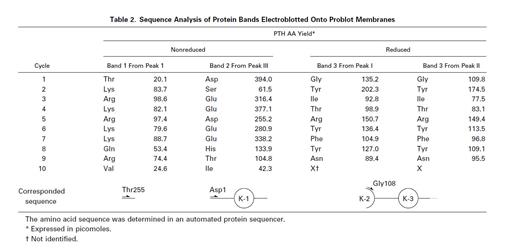
Peak I corresponded to three fragments starting with Thr 87, Gly 108, or Thr 255. SDS-PAGE of peak I showed two bands with molecular weight 35 kD and less than 14 kD (band 1) in the absence of reducing reagent (Fig 3B). Band 1 was found to have a molecular weight of about 5 kD by SDS-PAGE using Phast Gel High Density (data not shown). The amino acid sequence of band 1 was determined after blotting of the band on SDS-PAGE (Table 2). These results indicate that peak I was a mixture of two fragments; one was TFPI containing nicked K2, and the other was a C-terminal basic fragment starting from Thr 255 (band 1). A protein band of 35 kD in reduced gel was identified as a fragment starting at Gly 108 (Table 2), which corresponded to band 3 (Fig 1B). Peak II corresponded to two fragments starting with the N-terminal of the intact TFPI and Gly 108 (Table 1). SDS-PAGE of peak II showed a single band with molecular weight 43 kD in the absence of reducing reagent (Fig 3B). Under the reducing condition, peak II showed two major bands with molecular weights of 35 and 17 kD, respectively, and a minor band with molecular weight 43 kD. The band with molecular weight 35 kD was found to be band 3, since its amino acid sequence was identical to band 3 from peak I (Table 2). The 17-kD band and 43-kD band were identified as fragments of the N-terminal region of TFPI and intact TFPI, respectively. These results indicate that peak II contains TFPI with nicked K2 and a small amount of intact TFPI (and TFPI lacking the C-terminal region). Peak III was found to have the same N-terminal amino acid sequence as intact TFPI (Table 1). Peak III showed a single band on SDS-PAGE (band 2) with molecular weight 15 kD (Fig 3B), which corresponded to the elastase fragment of TFPI (residue no. 1-87).3 The amino acid sequence of band 2 was determined after blotting (Table 2).
Sites of hydrolysis identified from these analyses are shown in Table 1. These data demonstrate that thrombin cleaved TFPI at three peptide bonds: Lys 86-Thr 87 in the amino acid sequence between K1 and K2, Arg 107-Gly 108 (the reactive site for factor Xa in K2), and Lys 254-Thr 255 in the C-terminal basic region of TFPI. Since plasmin prefers lysine at the P1 position, the possibility of plasmin contamination in the thrombin preparation can be assumed. Therefore, we examined the effect of plasmin on rTFPI. We incubated rTFPI with plasmin at an enzyme to inhibitor ratio of 1:50 for 8 hours. However, degradation of rTFPI was not observed at all (data not shown).


Separation of thrombin digest of rTFPI on reverse phase-HPLC. (A) The 9.5 hours digest of rTFPI with thrombin was applied to a COSMOSIL 5C18-300 column that had been equilibrated with 0.05% trifluoroacetic acid. The fragments were eluted with 0.05% trifluoroacetic acid containing a linear concentration gradient of acetonitrile from 36% to 54%. (B) SDS-PAGE of each fraction, peak I, peak II, and peak III from reversed phase HPLC. See text for nomenclature of band 1, band 2, and band 3.
Separation of thrombin digest of rTFPI on reverse phase-HPLC. (A) The 9.5 hours digest of rTFPI with thrombin was applied to a COSMOSIL 5C18-300 column that had been equilibrated with 0.05% trifluoroacetic acid. The fragments were eluted with 0.05% trifluoroacetic acid containing a linear concentration gradient of acetonitrile from 36% to 54%. (B) SDS-PAGE of each fraction, peak I, peak II, and peak III from reversed phase HPLC. See text for nomenclature of band 1, band 2, and band 3.
We also examined the effect of this thrombin preparation on Boc-Val-Leu-Lys-MCA. Although plasmin cleaved the substrate significantly, this thrombin preparation did not cleave the substrate at all (data not shown).
To determine the order of priority of proteolysis by thrombin, SDS gels (Fig 1A and B) were analyzed by monitoring band 1, band 2, and band 3 using an image analyzing program. The increase of band 1 and band 2 in nonreducing gels indicated the cleavage of peptide bonds of Lys 254-Thr 255 and Lys 86-Asp 87, and the increase of band 3 in reducing gels, the cleavage of Arg 107-Gly 108 (Table 2). From the result of densitometric analysis of SDS-PAGE, it was proved that band 1 increased first, followed by band 3 and then band 2 (data not shown). This result indicated that thrombin first liberated the C-terminal fragment by cleaving Lys 254-Thr 255, and then cleaved Arg 107-Gly 108 in K2 and the Lys 86-Thr 87 bond between K1 and K2.
Incorporation of TFPI into fibrin clot and degradation by thrombin.To investigate the possible significance of TFPI cleavage by thrombin in vivo, TF/calcium ion and 125I-rTFPI were added to the plasma and TFPI degradation was examined by SDS-PAGE under the reducing condition. Figure 4 shows an autoradiograph of 125I-rTFPI in the serum and fibrin clot of normal and AT-depleted plasma. When TFPI degradation was compared in serum after addition of TF/calcium ion, TFPI was significantly degraded in AT-depleted plasma (lane 5), which was comparable to the degradation by thrombin (lane 2). However, degradation of TFPI in normal plasma (lane 4) was not as appreciable versus AT-depleted plasma. Degradation of TFPI in the fibrin clot also was not appreciable (lanes 6 and 7). Without TF/calcium ion, degradation of TFPI was not observed (lane 3). In the presence of 75 μmol/L argatroban, a specific thrombin inhibitor, degradation of TFPI was not observed (data not shown). These results indicate that degradation of TFPI by thrombin could be also observed in serum, but is markedly inhibited by AT.

Degradation of 125I-TFPI in serum and fibrin clot from normal or AT-depleted plasma. Radiolabeled rTFPI was mixed with plasma, and coagulation was induced by the addition of tissue factor and Ca2+, as described in Materials and Methods. After 20 hours, the serum and fibrin clot were separated and subjected to autoradiographic analysis. (Lane 1), intact rTFPI; (lane 2), rTFPI treated with thrombin; (lane 3), rTFPI incubated with normal plasma; (lane 4), rTFPI in serum from normal plasma; (lane 5), rTFPI in serum from AT-depleted plasma; (lane 6), rTFPI in fibrin clot from normal plasma; (lane 7), rTFPI in fibrin clot from AT-depleted plasma.
Degradation of 125I-TFPI in serum and fibrin clot from normal or AT-depleted plasma. Radiolabeled rTFPI was mixed with plasma, and coagulation was induced by the addition of tissue factor and Ca2+, as described in Materials and Methods. After 20 hours, the serum and fibrin clot were separated and subjected to autoradiographic analysis. (Lane 1), intact rTFPI; (lane 2), rTFPI treated with thrombin; (lane 3), rTFPI incubated with normal plasma; (lane 4), rTFPI in serum from normal plasma; (lane 5), rTFPI in serum from AT-depleted plasma; (lane 6), rTFPI in fibrin clot from normal plasma; (lane 7), rTFPI in fibrin clot from AT-depleted plasma.
Table 3 shows the results of experiments measuring recovery of radioactivity of TFPI from the clot and serum. After addition of TF/calcium ion to the plasma containing radiolabeled rTFPI, the serum and clot were separated by centrifugation after 15 minutes' and 20 hours' incubation. The clot was washed twice in the washing buffer. Table 3 shows that radioactivity in serum from normal plasma after 20 hours was higher than after 15 minutes, whereas radioactivity in the clot decreased. Radioactivity after both 15 minutes and 20 hours in serum from AT-depleted plasma was larger than in serum from normal plasma. The increase of radioactivity in the serum and the decrease in the clot from AT-depleted plasma were more prominent than those from normal plasma. Profiles of the increase and decrease of radioactivity from plasminogen-depleted plasma and from AT- and plasminogen-depleted plasma were indistinguishable versus normal plasma or AT-depleted plasma, respectively. These results, taken together with the results described in the previous section, demonstrate that a substantial portion of TFPI was degraded by thrombin after TF-induced coagulation and released into serum from the clot. The release of TFPI may not be mediated by plasmin, because degradation was observed to the same extent in plasminogen-depleted plasma and normal plasma. In these experiments, nonspecific binding of radiolabeled TFPI to the tube was 0.08% ± 0.01% (n = 3) of total radioactivity.
A notable amount of radioactivity was found in the clot (Table 3). After washing with TBS, 23.6% ± 3.8% (n = 3) of total radioactivity was washed out by the buffer containing 2.0 mol/L NaCl. However, radioactivity was not further washed out by 6 mol/L urea, and 17.3% ± 5.7% of the radioactivity initially used was found to be firmly associated with the clot. These results indicate that TFPI bound to fibrin in both a weakly associated form and a firmly associated form. When the association of rTFPI-C with fibrin was compared with that of rTFPI, the radioactivity of rTFPI-C in serum was higher and in fibrin lower, compared with that of rTFPI (Table 3). These results indicate that TFPI was incorporated into the fibrin clot mainly via the C-terminal basic region. Radioactivity in the clot from factor XIII–deficient plasma was not markedly different versus that from normal plasma, which indicates that factor XIII is not mainly involved in the association of TFPI with fibrin.
When fibrinogen was used instead of plasma and fibrin was formed by thrombin, incorporation of rTFPI into the fibrin clot was also observed (Table 4). In the presence of calcium ion or EDTA, about 30% of total radioactivity was found in the fibrin. After 20 hours' incubation, radioactivity in the supernatant was higher than after 15 minutes, while radioactivity in the clot decreased. When rTFPI-C was used, radioactivity was found mainly in the supernatant. Radioactivity in the supernatant after both 15 minutes' and 20 hours' incubation was about 80% of the total radioactivity. Figure 5A shows the effect of C-terminal basic peptide on rTFPI binding to the fibrin clot. With the increase of C-terminal peptide, binding of rTFPI to the fibrin clot was decreased, whereas radioactivity in the supernatant was increased. Figure 5B shows the effect of Gly 212-Phe 243 peptide on rTFPI binding to the fibrin clot. In the presence of 300 μmol/L of the peptide, incorporation of TFPI into the fibrin clot was not affected. When nonspecific binding of rTFPI to the tube (4.34% ± 0.21% of total radioactivity after 15 minutes' incubation, n = 3) was taken into account, rTFPI binding to fibrin was observed to be almost completely inhibited in the presence of 300 μmol/L of the C-terminal peptide.

Effects of TFPI binding to fibrin clot by C-terminal basic peptide (A) or Gly 212-Phe 243 peptide in the K3 domain (B) of TFPI. Various concentrations of each peptide were mixed with fibrinogen, and radiolabeled TFPI was added. Fibrin was formed by the addition of 2 μg/mL of thrombin. After 15 minutes, fibrin was removed and washed by TBS. The radioactivity of the supernatant and the washed fibrin was then measured. Each bar represents the mean ± SD from four samples. (□), Supernatant; (▪), fibrin clot.
Effects of TFPI binding to fibrin clot by C-terminal basic peptide (A) or Gly 212-Phe 243 peptide in the K3 domain (B) of TFPI. Various concentrations of each peptide were mixed with fibrinogen, and radiolabeled TFPI was added. Fibrin was formed by the addition of 2 μg/mL of thrombin. After 15 minutes, fibrin was removed and washed by TBS. The radioactivity of the supernatant and the washed fibrin was then measured. Each bar represents the mean ± SD from four samples. (□), Supernatant; (▪), fibrin clot.
When 125I-TFPI was incubated with 10 or 100 U/mL heparin solution for 30 minutes at 37°C and then with fibrinogen and thrombin, TFPI incorporation was not affected by heparin. Since heparin binds to TFPI via the C-terminal part of TFPI, the inhibition of incorporation could be expected. As shown in the helical wheel diagram of the C-terminal part of TFPI (250-268),17 the basic residues in this region could be divided into two sides. One of them is expected to react with heparin. It may be expected that another side of the helical face of this helical wheel diagram is concerned with the interaction between TFPI and fibrin. It is also possible that the interaction site of TFPI with fibrin exists in the sequence between Gly 242 and IIe 253.
These results indicate that rTFPI was incorporated into fibrin mainly via its C-terminal basic region, which was not mediated by factor XIIIa, and that the fibrin-bound TFPI was released into solution by thrombin. Since these results were derived from experiments using radiolabeled TFPI, we examined whether TFPI in plasma will be associated with fibrin after coagulation, using ELISA for TFPI antigen. Coagulation of human normal plasma was induced by addition of TF and calcium ion, and the serum was separated as already described. The free-form TFPI antigen level in the serum was 5.6% of the level in normal plasma (n = 2). This result suggests that TFPI in plasma will be associated with fibrin on in vivo thrombosis and then degraded by fibrin-bound thrombin as already demonstrated.
DISCUSSION
In this study, we demonstrated that thrombin reduced the inhibitory activity of rTFPI by cleaving three peptide bonds, Lys 254-Thr 255 in the C-terminal basic region, Arg 107-Gly 108 (reactive site toward factor Xa in K2), and Lys 86-Thr 87 between K1 and K2, in that order. These results were accomplished by amino acid sequence analysis of fragments of thrombin digest and by SDS-PAGE analysis. As previously reported,7,8,18,19 cleavage of a peptide bond between K1 and K2 of TFPI or liberation of the C-terminal basic region of TFPI reduced the inhibitory activity of TFPI. Cleavage of the reactive site toward factor Xa in K2 of TFPI evidently leads to the loss of inhibitory activity of TFPI. Therefore, it is reasonable to conclude that the inhibitory activity of TFPI was reduced by thrombin after cleavage of either one of the three peptide bonds. As previously reported,20 there are two major groups at Arg/Lys-Xaa bonds that are susceptible to thrombin cleavage: those with P2-P4 residues as hydrophobic amino acids, and those with P1′ or P2 residues as Gly. Three scissile peptide bonds on TFPI are consistent with the thrombin specificity. In the presence of heparin, thrombin did not cleave Lys 254-Thr 255 in the C-terminal basic region of TFPI, which is supposed to be due to the binding of heparin to the basic region. Calcium ion or thrombomodulin did not affect the profile of TFPI degradation by thrombin.
As previously reported21,22 and as confirmed by the present study, thrombin activity was not influenced by an excess of TFPI. Therefore, it is not surprising that TFPI could be a substrate for thrombin. However, it should be discussed how thrombin cleaved the reactive site toward factor Xa in K2, which is resistant to cleavage by factor Xa. TFPI inhibited trypsin (Ki, 0.63 nmol/L)1 and plasmin (Ki, 15.0 nmol/L),23 in addition to factor Xa (Ki, 26.4 pmol/L).24 All these proteases have Gln at position 192 (in the chymotrypsin numbering system25 ), whereas thrombin has Glu at the same position. Glu 192-Gln substitution of thrombin resulted in inhibition of the mutant protease by TFPI via K2.20 As demonstrated in the trypsin-BPTI complex, Gln 192 of trypsin was involved in hydrogen bonding with several residues of BPTI and a water molecule in the active site.26,27 It has been speculated that a conformation of the primary contact region of the inhibitor is supported by a secondary contact region in the Kunitz-type inhibitor complex. Hydrogen bonds and disulfide bonds between the primary contact region and the secondary contact region and hydrogen bonds between an enzyme and the primary contact region may contribute to formation of the stable complex.28-31 Considering all these results and speculations, we suggest that TFPI did not form a stable complex with thrombin and was therefore cleaved by thrombin, since Glu 192 in thrombin prevented effects on the hydrogen-bonded structures of the active site or the primary contact region and, as a consequence, maintained the conformation of these regions. Another feature of thrombin is the presence of a unique Trp-containing surface loop at each side of the active site (Trp 60D and Trp 148 loop).25,32 Modeling studies on the interaction of BPTI with thrombin predicted collision of several residues in the inhibitor, primarily Tyr 35′, with residues in the 60-loop of thrombin, primarily Trp 60D, which would result in steric hindrance to prevent complex formation.33 From the results presented here, it seems that both the Trp loop and Glu 192 of thrombin contributed to make thrombin accessible to the Arg 107-Gly 108 bond in K2 of TFPI.
As shown in the present study, TFPI was degraded by thrombin itself and not by other proteases, particularly not plasmin. In the purified system, degradation of TFPI by thrombin was completely inhibited in the presence of thrombin inhibitors. The possibility of plasmin contamination in the thrombin preparation was excluded by the fact that the preparation had no ability to hydrolyze plasmin substrate and that plasmin was inhibited by TFPI and did not cleave TFPI. Degradation was more prominent in AT-depleted plasma than in normal plasma. However, degradation was similar in plasminogen-depleted plasma versus normal plasma. These results indicate that thrombin, not plasmin, was the protease responsible for degradation of TFPI, although the degradation rate was markedly inhibited by AT in plasma.
It was observed that a substantial amount of radioactivity was found in the fibrin clot and that radioactivity in the serum was increased after 20 hours' incubation compared with 15 minutes' incubation. The association of rTFPI-C with the fibrin clot was weaker than that of rTFPI. These results indicated that TFPI was initially incorporated into the fibrin clot and released after degradation with thrombin. This speculation was strongly supported by the result of the more simple experimental condition, ie, the mixture of radiolabeled rTFPI, fibrinogen, and thrombin. Similar results that TFPI was incorporated into fibrin and released after prolonged incubation were obtained in the presence of calcium ion or EDTA. Incorporation was completely prevented by a synthetic C-terminal basic peptide of TFPI. These two lines of evidence demonstrated that TFPI was associated with fibrin during coagulation and degraded by fibrin-bound thrombin. It has been established that fibrin enhances enzyme reactions by binding with substrate and enzyme, such as thrombin-mediated activation of factor XIII and plasminogen activator–mediated activation of plasminogen.34,35 In these reactions, both substrate and enzyme bind to the specific binding sites of fibrin, and the collisions of the reactants are enhanced by formation of the ternary complex.36-39 Although we did not identify the specific binding site of fibrin for TFPI, we show that the degradation of TFPI by fibrin-bound thrombin was enhanced by the formation of the complex on fibrin, as in the case of activation of factor XIII or plasminogen. The results also indicated that TFPI was bound to fibrin via the C-terminal basic region of TFPI, which was not mediated by factor XIIIa. Many proteins are reported to bind to fibrin by a covalent bond mediated by factor XIIIa, such as α2-plasmin inhibitor40 and plasma fibronectin,41 and by a noncovalent bond, such as plasminogen42 and tissue-type plasminogen activator.43 Several lines of evidence suggest that the association of TFPI with fibrin is mediated by ionic interaction, not by covalent bonding by factor XIIIa. Although the interaction sites of fibrin for TFPI remain to be established, our present results indicate that the cluster of basic amino acid residues in the C-terminal region of TFPI interacted with the cluster of negatively charged residues in fibrin. The finding that about 40% of the radioactivity bound to fibrin was not washed by 6 mol/L urea indicates that TFPI was associated with fibrin both by weak and strong noncovalent binding. We propose that incorporation of TFPI into the fibrin clot occurred during or after formation of fibrin, because we could not establish TFPI binding to fibrinogen (Ohkura et al, unpublished observations, 1996).
From the evidence reported thus far, it has been speculated that endothelial cell–associated TFPI plays an important role in the anticoagulant property of endothelial cells, binding to proteoglycans on the cells. It has also been shown that TFPI is stored intracellularly in endothelial cells and redistributes to the surface of the cells on stimulation with thrombin.44 This pathway is believed to be a negative-feedback control of blood coagulation. TFPI in plasma is cleared by low-density lipoprotein receptor–related protein in hepatoma cells.45,46 In addition, cell-surface heparan sulfate proteoglycans46,47 associated with sinusoidal cells in the liver have been proposed to be involved in the clearance of TFPI. It is evident that fibrin-bound thrombin takes part in the regulation of thrombosis in vivo, as established in many other studies. During the conversion of fibrinogen to fibrin, thrombin is removed from solution and bound to fibrin.48-50 The in vivo half-life of thrombin and its concentration in circulation are very small. However, the fibrin clot can serve as a reservoir of enzymatically active thrombin. Thrombin has been shown to be associated with fibrin through a site distinct from its catalytic center, and to retain its enzymatic activity.49-52 Inactivation of fibrin-bound thrombin by antithrombin occurs more slowly than in plasma.48 In our experiments, cleavage and inactivation of TFPI by thrombin was relatively slow, but it is possible that the fibrin-bound TFPI can be exposed to active thrombin for a long time on the fibrin clot. Furthermore, we have shown that free-form TFPI in plasma markedly decreased and associated with fibrin after coagulation of plasma. Thus, the present findings indicate that TFPI is incorporated into fibrin after in vivo coagulation and inactivated by the fibrin-bound thrombin, degrading into the aqueous phase. Although the presence of truncated forms of TFPI in plasma has been suggested, the mechanism of truncation is not clear. Degradation of TFPI by thrombin may be related to these truncated forms of TFPI. TFPI truncation that leads to reduced TFPI activity may be more significant in various thrombotic diseases. The novel degradation pathway of TFPI proposed in the current study may give new insight into the mechanism of TFPI clearance, the regulation of its catabolism, and the role of TFPI in thrombosis and hemostasis.
ACKNOWLEDGMENT
We express our thanks to Dr Toshiyuki Miyata and Dr Koichi Kokame, National Cardiovascular Center Research Institute, for helpful suggestions during this study. We thank Professor J. Evan Sadler, Washington University School of Medicine, St Louis, for comments on the study. We also thank Professor Michio Matsuda, Institute of Hematology, Jichi Medical School, and Dr Gilbu Soe, Central Research Laboratories, Iatron Laboratories Inc, for helpful comments and discussions.
Supported in part by Special Coordination Funds for Promoting Science and Technology (Encouragement System of Center of Excellence [COE]) from the Science and Technology Agency of Japan.
Address reprint requests to Hisao Kato, PhD, National Cardiovascular Center Research Institute, Fujishirodai 5-7-1, Suita, Osaka 565, Japan.