

Abstract
Dendritic-cell (DC) and natural killer (NK)–cell interactions are critical in sculpting the adaptive immune response. However, the mechanisms by which DCs down-regulate NK-cell functions are not well understood. NK-cell function is inhibited by transforming growth factor beta (TGF-β), but DCs do not appear to produce TGF-β. We have previously shown that activated human DCs produce large amounts of activin-A, a TGF-β superfamily member, which autoregulates DC function. The present report shows that NKcells express type I and II activin receptors and that activin-A triggers NK-cell Smad 2/3 signaling. Furthermore, activin-A directly regulates NK cell functions by (1) down-regulating the T-box transcription factor T-bet and interferon gamma (IFN-γ) but not perforin or granzyme mRNA; (2) suppressing NK-cell IFN-γ production as potently as TGF-β; and (3) suppressing NK-cell CD25 expression and proliferation and sculpting NK-cell cytokine and chemokine profiles. Interestingly, unlike TGF-β, activin-A weakly down-regulates the NK-cell natural cytotoxicity receptors (NCRs) NKp30 and NKG2D but does not attenuate their cytotoxic function. These findings provide the first evidence for a novel immune regulatory role of activin-A during DC-mediated NK-cell regulation, highlighting the potential of antagonizing activin-A signaling in vivo to enhance NK cell–mediated immune functions and adaptive immunity.
Introduction
Natural killer (NK) cells are innate immune cells specialized in the recognition and killing of virus-infected or tumor cells.1-3 NK cells also play an important role in the regulation of both innate and adaptive immunity via direct interaction with dendritic cells (DCs) in the lymph node or within inflamed tissues via production of interferon (IFN)–γ.4 DCs detect pathogens via an array of pattern-recognition receptors5 and pathogen encounter induces DC maturation and production of proinflammatory chemokines and cytokines such as interferon-induced protein (IP)–10, interleukin (IL)–8, IL-6, tumor necrosis factor α (TNF-α), IFN-α, and IL-12,6,7 which attract and activate innate effector cells.8,9
DC-derived IL-12p70 can act as a potent cofactor for enhancing NK-cell cytotoxicity and IFN-γ production, which is responsible for the initial shaping of T-helper type 1 (Th-1) immunity.10,11 We recently have shown that as well as producing IL-12p70, activated DCs also rapidly secrete high levels of activin-A after exposure to bacteria, specific Toll-like receptor (TLR) ligands, or CD40 ligand (CD40L).12 Furthermore, DC-derived activin-A acts as a potent autocrine regulator of DC secretion of IL-12p70 as well as other proinflammatory chemokines (IL-8, IP-10, and monocyte chemoattractant protein [MCP]–1) and cytokines (IL-6, IL-10, and TNF-α).12
Activin-A is a transforming growth factor (TGF)–β superfamily member and shares with TGF-β the Smad intracellular signaling proteins.13 Activin-A is separate and distinct from TGF-β and binds activin type I (ALK 2, 4, or 7) and type II (ActRIIA and ActRIIB) receptors.14 Activin-A is also released more rapidly than TGF-β during acute systemic inflammation and the signaling involves unique serine threonine kinase receptor subunits.14
Activin's ability to stimulate the release of follicle-stimulating hormone (FSH) was its first described biological property.15 However, activin's pleiotropic nature has now become more fully understood. It has been found to have both pro-16 and antiproliferative17,18 effects on tumor cells as well as involvement in disease states such as pancreatic fibrosis,19 rheumatoid arthritis,20 and diabetes.21 More recently activin-A has been described as an essential growth factor involved in embryonic stem cell renewal and pluripotency.22,23
Activin-A's biological effects are controlled through interaction with its naturally occurring high-affinity antagonist, follistatin.24-26 Two follistatin molecules neutralize the actions of one activin-A molecule through antagonism of both type I and type II activin receptor binding sites.27 During inflammation, systemic release of follistatin occurs after the release of activin-A, and its actions are thought to modulate and suppress activin's effects.28
With activated DCs producing such large amounts of activin-A12 and with the well-documented interaction between DCs and NK cells,4 we hypothesized that activin-A may influence NK-cell function. Our studies demonstrate that NK cells express receptors for activin-A and that activin-A attenuates NK-cell IFN-γ production, proliferation, and phenotypic maturation but has no effect on their capacity to kill tumor cell targets. This novel immune-regulatory effect of activin-A has important implications for our understanding of immune regulation and for the development of strategies to manipulate NK-cell function in vivo.
Methods
Cell culture
Peripheral blood mononuclear cells (PBMCs) from buffy coats of healthy donors (Red Cross Blood Bank, Melbourne, Australia) were prepared by Ficoll-Paque density gradient centrifugation (GE Healthcare, Little Chalfont, United Kingdom). Use of these cells was granted ethical approval by the institutional review board of Austin Health Hospital. NK cells were isolated with the use of a human NK-cell isolation kit (Miltenyi Biotec, Auburn, CA). Monocytes were isolated by positive selection with the use of magnetic beads specific for CD14 (Miltenyi Biotec). Monocyte-derived DCs (MoDCs) were generated by culturing CD14+ cells with granulocyte monocyte colony-stimulating factor (GM-CSF) and IL-4 for 6 to 7 days. Cell cultures were maintained in RPMI 1640 (Sigma-Aldrich, St Louis, MO) supplemented with 20 mmol/L HEPES, 60 mg/L penicillin, 12.5 mg/L streptomycin, 2 mmol/L l-glutamine, 1% nonessential amino acids, and 10% heat-inactivated fetal calf serum (FCS). MoDCs were plated in fresh culture media supplemented with GM-CSF. NK cells were cultured in 20 U/mL IL-2 and stimulated with the following conditions: 10 ng/mL IL-12 or IL-12 together with 1 μg/mL of the synthetic lipopeptide (S)-(2,3-bis(palmitoyloxy)-(2RS)-propyl)-N-palmitoyl-(R)-Cys-(S)-Ser(S)-Lys4-OH trihydrochloride (Pam3Cys), 12.5 μg/mL polyriboinosinic:polyribocytidylic acid (poly I:C; InvivoGen, San Diego, CA), or 100 ng/mL Escherichia coli–derived lipopolysaccharide (LPS; Sigma-Aldrich); which bind TLR 2/6, 3, and 4, respectively. A total of 20 ng/mL GM-CSF was added to NK-cell and DC cocultures.
Quantitative real-time polymerase chain reaction
RNA was isolated from NK cells with the use of the RNeasy Mini Kit (QIAGEN, Hilden, Germany), and cDNA was synthesized as previously described.29 A total of 1 μL cDNA was used as template for quantitative real-time polymerase chain reaction (RT-PCR). Gene expression levels were quantified with the use of a Stratagene Mx3005P machine (La Jolla, CA). Primers were designed for detection of ALK4, ActRIIA and B, TGF-βRI and TGF-βRII, T-bet, IFN-γ, perforin, granzyme A and B, and IL-12RBI and IL-12RBII mRNA and used in combination with probes from a Universal ProbeLibrary (Roche Applied Science, Indianapolis, IN). 18S rRNA Pre-Developed TaqMan Assay Reagents (PDARs; Applied Biosystems, Foster City, CA) were used for normalization. PCRs were set up in 96-well plates and analyzed with the use of Mx Pro version 3.2 (Stratagene). The frequency of target gene expression in relation to 18S was calculated as 2(Ct18S − Ct target gene). Relative expression also was calculated by the use of the ΔCt method and was expressed relative to a calibrator, in this case, ex vivo–purified NK cells or relevant NK-cell control cultures:

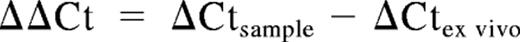
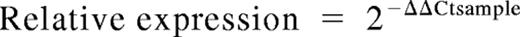
The following qRT-PCR primers were used: ALK4, (F) gacgctccaggatcttgtct and (R) gtgcgctggacaaagagg; ActRIIA, (F) gactttgggttggccttaaa and (R) gtacctccgggtaccaacct; ActRIIB, (F) tgtcaagatcttcccactcca and (R) catgccaggtgtgctgaa; TGF-βRI, (F) aaattgctcgacgatgttcc and (R) cataataaggcagttggtaatcttca; TGF-βRII, (F) gggaaatgacatctcgctgta and (R) caccttggaaccaaatggag; T-bet, (F) tccaagtttaatcagcaccaga and (R) tgacaggaatgggaacatcc; IFN-γ, (F) ggcattttgaagaattggaaag and (R) tttggatgctctggtcatctt; perforin, (F) ccgcttctctatacgggattc and (R) gcagcagcaggagaaggat; granzyme A, (F) ttaaccctgtgattggaatgaat and (R) agggcttccagaatctccat; granzyme B, (F) cggtggcttcctgatacaa and (R) ccccaaggtgacatttatgg; IL-12RBI, (F) cggctgaccctgaaagag and (R) cagcccttgacagccttc; and IL-12RBII, (F) gactcagatatcggcccagt and (R) tgtcttcctttggcctttgta.
Western immunoblot analysis
NK cells were plated in culture media supplemented with 20 U/mL IL-2 alone or with the addition of 50 ng/mL recombinant activin-A (R&D Systems, Minneapolis, MN). Smad 2/3 and β-actin were measured in samples with the use of specific antibodies according to the manufacturer's instructions (Cell Signaling Technology, Danvers, MA).
Cytokine enzyme-linked immunosorbent assay, multiplex cytokine, and chemokine arrays
Cytokine enzyme-linked immunosorbent assay (ELISA) kits (BD OptEIA; BD Biosciences, San Diego, CA) were used to quantify IFN-γ. Cytokine and chemokine bead arrays (LINCOplex) were used to quantify IFN-γ, IL-1β, IL-6, IL-8, IL-10, TNF-α, GM-CSF, IP-10, MCP-1 and macrophage inflammatory protein (MIP)–1α and β, and samples were analyzed on a Luminex instrument (all Millipore, St Charles, MO).
Assessment of cell viability
Purified NK cells were cultured in media supplemented with IL-2 plus IL-12 with or without increasing doses of activin-A or TGF-β (PeproTech, Rocky Hill, NJ). Polycaspase, caspase 3/7 or 8 was detected with fluorochrome inhibitor of caspases (FLICA), according to the manufacturer's instructions (ImmunoChemistry Technologies, Bloomington, MN) and measured by flow cytometry. Propidium iodide (PI) was used to detect dead cells.
Assessment of NK-cell proliferation
Purified NK cells were labeled with 1 μmol/L 5-(and 6)-carboxyfluorescein diacetate succinimidyl ester (CFSE; Molecular Probes/Invitrogen, Carlsbad, CA) at 37°C for 10 minutes before washing 3 times in cold RPMI. Labeled cells were cultured with cytokines and TLR ligands with or without the addition of 100 ng/mL activin-A for 5 days. CFSE intensity was measured on CD56+ cells by flow cytometry.
Assessment of NK-cell cytotoxicity and measurement of CD69, NKp30, and NKG2D expression
Purified NK cells were cultured for 3 days with cytokines with or without the addition of 100 ng/mL activin-A or TGF-β. K562 cells, an erythroleukemia cell line, or Jurkat cells, a human T-cell leukemia line, were labeled with 15 μmol/L calcein (Molecular Probes) according to the manufacturer's instructions. Washed NK cells and labeled targets were cocultured at the indicated ratios for 4 hours and percent killing determined by measurement of calcein dye release into cell supernatants. NK cells were also cryopreserved ex vivo or cryopreserved after the 3-day culture period and then thawed, washed, and costained with anti–human CD56, CD69, NKp30, and NKG2D antibodies (BD Biosciences, San Diego, CA). Expression was determined by flow cytometry.
Effects of binding activin-A in vitro
To assess the effects of blocking activin-A signaling, SB431542 (Sigma-Aldrich), a selective inhibitor of the activin type I receptor ALK-4 was added to purified NK-cell cultures or recombinant human follistatin (FS-300; R&D Systems) was added to NK-cell and DC-cell cocultures.
Statistics
Unless otherwise indicated, results are expressed as the mean plus or minus one SD of 3 or more donors. P values less than .05 are considered significant.
Results
Activin and TGF-β receptor gene regulation and Smad signaling in human NK cells
To assess the relevance of the activin system to human NK cells, the expression pattern of the type I and II human activin and TGF-β receptors mRNAs was assessed by qRT-PCR in NK cells purified from blood. The analysis demonstrated that highly purified human NK cells (Figure S1, available on the Blood website; see the Supplemental Materials link at the top of the online article) coexpress mRNA for the activin type I (ALK4) and II (ActRIIA and ActRIIB) and the TGF-β type I (RI) and II (RII) receptors ex vivo (Figure 1A,B). Culture of NK cells for 16 hours in media containing IL-2 and IL-12 resulted in a 3- to 4-fold increase in ALK4, ActRIIA, and ActRIIB mRNA levels (Figure 1C). Addition of recombinant activin-A or TGF-β to these cultures resulted in a further increase in ALK4 mRNA levels but had no further effect on ActRIIA or ActRIIB expression (Figure 1C). Conversely, the expression of the type I and II TGF-β receptor mRNAs were unaltered by culture in media containing IL-2 and IL-12, but addition of activin-A or TGF-β resulted in significant down-regulation of expression (Figure 1D). Finally, the addition of recombinant activin-A to ex vivo purified NK cells resulted in enhanced intracellular Smad 2/3 levels within 4 hours (Figure 1E). Together these results demonstrate that human NK cells express receptors for and respond to activin-A and prompted further investigations into the potential roles of activin-A signaling in NK-cell function.

Activin-A and TGF-β receptor expression and activin-A–induced Smad signaling in NK cells. NK cells were purified from buffy coats of healthy donors and assessed for expression of activin type I (ALK4) and type II (ActRIIA and ActRIIB) or TGF-β type I (RI) and type II (RII) receptors mRNA by qRT-PCR (A-D). In brief, NK cells were lysed immediately after purification or after 16 hours of culture in media supplemented with IL-2 (20 U/mL) + IL-12 (10 ng/mL) with or without the addition of recombinant activin-A or TGF-β. RNA was then extracted and qRT-PCR performed. (E) Western blotting for detection of Smad 2/3 or β-actin expression. In brief, purified NK cells (5 × 105) were cultured in 24-well plates for 4 hours with either IL-2 alone or with IL-2 and recombinant human activin-A (50 ng/mL). NK cells were then lysed and protein extracted. Data are shown as either one representative experiment from 3 separate donors (E) or the mean plus or minus 1 SD of 3 separate donors (A-D). *P < 0.01 and **P < .02 versus ex vivo levels.
Activin-A and TGF-β receptor expression and activin-A–induced Smad signaling in NK cells. NK cells were purified from buffy coats of healthy donors and assessed for expression of activin type I (ALK4) and type II (ActRIIA and ActRIIB) or TGF-β type I (RI) and type II (RII) receptors mRNA by qRT-PCR (A-D). In brief, NK cells were lysed immediately after purification or after 16 hours of culture in media supplemented with IL-2 (20 U/mL) + IL-12 (10 ng/mL) with or without the addition of recombinant activin-A or TGF-β. RNA was then extracted and qRT-PCR performed. (E) Western blotting for detection of Smad 2/3 or β-actin expression. In brief, purified NK cells (5 × 105) were cultured in 24-well plates for 4 hours with either IL-2 alone or with IL-2 and recombinant human activin-A (50 ng/mL). NK cells were then lysed and protein extracted. Data are shown as either one representative experiment from 3 separate donors (E) or the mean plus or minus 1 SD of 3 separate donors (A-D). *P < 0.01 and **P < .02 versus ex vivo levels.
Dendritic cell–derived activin-A regulates NK cell IFN-γ production
NK cell–derived IFN-γ is a potent factor involved in shaping Th1 immunity.11 To investigate whether DC-derived activin-A could directly regulate NK cell IFN-γ production, we cocultured MoDCs together with autologous NK cells and stimulated the cells with cytokines and poly I:C. We have shown previously that poly I:C stimulates MoDCs to produce large amounts of activin-A and that this activin-A can be neutralized by the addition of follistatin.12 We found that IFN-γ was not detected in cultures of immature MoDCs alone that were stimulated with IL-12 and poly I:C nor in cultures of immature MoDCs mixed with NK cells (Figure 2A). However, coculture of immature MoDCs and NK cells in the presence of IL-12 and poly I:C resulted in IFN-γ production by the NK cells and, most importantly, this was significantly increased by addition of the activin-A antagonist, follistatin (Figure 2A). Our previous analysis demonstrated that NK cells do not produce activin-A12 and addition of follistatin to cytokine-stimulated NK cells had no effect on their IFN-γ production (Figure 2B), excluding this as a simplistic explanation for the observed enhancing effects of follistatin in the coculture experiments.

The effects of blocking DC-derived activin-A with follistatin upon NK-cell IFN-γ production. Immature MoDCs were cultured at 5 × 104 cells in 96-well plates in media supplemented with IL-2 (20 U/mL) and GM-CSF (20 ng/mL) either alone or together with 105 autologous NK cells for 24 hours. (A) IFN-γ production in cocultures of MoDCs with various combinations (as shown) of IL-12 (10 ng/mL) and poly I:C (12.5 μg/mL) and/or NK cells and/or follistatin (400 ng/mL). (B) IFN-γ production by NK cells cultured with IL-2 and IL-12 with or without follistatin (400 ng/mL) for 24 hours. Data represent the mean plus or minus 1 SD of 3 separate donors. **P < .05 versus no follistatin.
The effects of blocking DC-derived activin-A with follistatin upon NK-cell IFN-γ production. Immature MoDCs were cultured at 5 × 104 cells in 96-well plates in media supplemented with IL-2 (20 U/mL) and GM-CSF (20 ng/mL) either alone or together with 105 autologous NK cells for 24 hours. (A) IFN-γ production in cocultures of MoDCs with various combinations (as shown) of IL-12 (10 ng/mL) and poly I:C (12.5 μg/mL) and/or NK cells and/or follistatin (400 ng/mL). (B) IFN-γ production by NK cells cultured with IL-2 and IL-12 with or without follistatin (400 ng/mL) for 24 hours. Data represent the mean plus or minus 1 SD of 3 separate donors. **P < .05 versus no follistatin.
Activin-A suppresses cytokine and TLR-3-mediated NK cell IFN-γ production
Although the aforementioned experiments established a regulatory role for DC-derived activin-A during NK-cell and DC interactions, its neutralization in this context does not allow distinction between activin's potential autocrine effects on the DCs or paracrine effects on the NK cells. Therefore, to test whether NK-cell production of IFN-γ was influenced by activin-A in the absence of DCs, we cultured human NK cells with IL-2 and IL-12 together with increasing concentrations of recombinant activin-A or TGF-β and measured IFN-γ levels in culture supernatants after 24 hours. We found that activin-A was as potent as TGF-β in the inhibition of NK-cell IFN-γ production (Figure 3A). Under these conditions, activin-A also significantly inhibited mRNA expression levels of the transcription factor T-bet and decreased expression of IFN-γ (Figure 3B).

The effects of activin-A and TGF-β on NK-cell IFN-γ production, gene regulation, and viability. Purified NK cells were cultured at 105 (A,E) or 106 (B-D) in 96- or 24-well plates, respectively, in media supplemented with IL-2 (20 U/mL) and IL-12 (10 ng/mL) in the absence or presence of increasing concentrations of activin-A or TGF-β. (A) NK-cell production of IFN-γ by ELISA after 24 hours. (B-D) Detection of T-bet, IFN-γ, Perforin, Granzyme A and B, IL-12RBI and IL-12RBII mRNA expression by qRT-PCR ex vivo or after 16 hours of stimulation in the absence of presence of activin-A or TGF-β (each 100 ng/mL). (E) Detection of cell viability by PI and apoptosis by poly-caspase, caspase 3/7, and caspase 8 expression using flow cytometry. Data represent the mean plus or minus 1 SD of 3 separate donors. *P < .01 and **P < .05 versus no addition of activin-A or TGF-β or versus ex vivo– or cytokine-stimulated levels.
The effects of activin-A and TGF-β on NK-cell IFN-γ production, gene regulation, and viability. Purified NK cells were cultured at 105 (A,E) or 106 (B-D) in 96- or 24-well plates, respectively, in media supplemented with IL-2 (20 U/mL) and IL-12 (10 ng/mL) in the absence or presence of increasing concentrations of activin-A or TGF-β. (A) NK-cell production of IFN-γ by ELISA after 24 hours. (B-D) Detection of T-bet, IFN-γ, Perforin, Granzyme A and B, IL-12RBI and IL-12RBII mRNA expression by qRT-PCR ex vivo or after 16 hours of stimulation in the absence of presence of activin-A or TGF-β (each 100 ng/mL). (E) Detection of cell viability by PI and apoptosis by poly-caspase, caspase 3/7, and caspase 8 expression using flow cytometry. Data represent the mean plus or minus 1 SD of 3 separate donors. *P < .01 and **P < .05 versus no addition of activin-A or TGF-β or versus ex vivo– or cytokine-stimulated levels.
To extend the analysis, we measured the effects of activin-A and TGF-β on the expression of the NK-cell cytolytic molecules perforin, granzyme A and B, together with expression of the IL-12BI and BII receptors. First, the analysis demonstrated that ex vivo–purified NK cells expressed moderate mRNA levels for granzyme A and IL-12RI but high levels of perforin and granzyme B and very high levels of IL-12RBII (data not shown). Culture in IL-2 and IL-12 did not effect perforin expression but up-regulated granzyme A and granzyme B mRNA expression (Figure 3C). However, the extent of this up-regulation varied considerably between donors and ranged from 2- to 200- and 3- to 10-fold, respectively (Figure 3C). The addition of activin-A did not affect either perforin or granzyme A or B expression whereas TGF-β specifically down-regulated granzyme B expression (Figure 3C). Culture in IL-2 and IL-12 increased IL-12RBI mRNAs, which were decreased by TGF-β but not affected by activin-A (Figure 3D). Conversely, there was a modest down-regulation of IL-12RBII mRNA levels upon culture in IL-2 and IL-12, but as with IL-12BI receptor mRNA, these levels were further decreased by TGF-β but not significantly affected by activin-A (Figure 3D). Taken together, these results demonstrate important differences in NK-cell gene regulation induced by TGF-β or activin-A exposure. Finally, neither activin-A nor TGF-β induced apoptosis of NK cells under these conditions (Figure 3E), excluding this as a simplistic explanation for the IFN-γ suppression observed.
The addition of poly I:C or LPS, but not Pam3Cys, to IL-2- and IL-12-stimulated NK-cell cultures resulted in enhanced NK-cell IFN-γ production (Figure S2A), and this was also suppressed by the addition of activin-A (Figure 4A and data not shown). Activin-A also significantly suppressed expression of the IL-2 receptor α-chain, CD25, on cytokine and TLR-3–stimulated NK cells (Figure S2B). Importantly, the suppression of NK-cell IFN-γ secretion by activin-A could be reversed by addition of the activin-A type I receptor antagonist, SB431542 (Figure 4B).

The effects of activin-A on NK-cell IFN-γ production. Purified NK cells were cultured at 105 in 96-well plates in media supplemented with IL-2 (20 U/mL) alone or IL-2 + IL-12 (10 ng/mL) with or without the addition of poly I:C (12.5 μg/mL) in the absence or presence of increasing concentrations of activin-A. (A) NK-cell production of IFN-γ by ELISA. (B) NK-cell production of IFN-γ by ELISA after pretreatment with the activin type I receptor antagonist SB431542 (10 μmol/L) for 30 minutes before addition of activin-A. Data represents the mean plus or minus 1 SD of 3 separate donors. *P < .01 versus no activin-A and versus SB431542 pretreatment.
The effects of activin-A on NK-cell IFN-γ production. Purified NK cells were cultured at 105 in 96-well plates in media supplemented with IL-2 (20 U/mL) alone or IL-2 + IL-12 (10 ng/mL) with or without the addition of poly I:C (12.5 μg/mL) in the absence or presence of increasing concentrations of activin-A. (A) NK-cell production of IFN-γ by ELISA. (B) NK-cell production of IFN-γ by ELISA after pretreatment with the activin type I receptor antagonist SB431542 (10 μmol/L) for 30 minutes before addition of activin-A. Data represents the mean plus or minus 1 SD of 3 separate donors. *P < .01 versus no activin-A and versus SB431542 pretreatment.
To extend the analysis, supernatants generated from the aforementioned experiments were screened with the use of a multiplex cytokine and chemokine array system. This method first confirmed the IFN-γ ELISA data by showing activin's suppressive effects but also demonstrated that IFN-γ (6-12 ng/mL) was the most abundant cytokine produced by activated NK cells (Figure 5A). Moreover, the array data demonstrated that activin-A also suppressed NK-cell production of IL-6, TNF-α, GM-CSF, and IL-1β, which were the next most abundantly produced cytokines, respectively (Figure 5A). Interestingly, activin-A significantly increased the low amounts (10-30 pg/mL) of IL-10 produced by NK cells (Figure 5A). Furthermore, activin-A significantly suppressed NK-cell production of MIP-1β (400-700 pg/mL), and also suppressed MIP-1α, IL-8, and IP-10, which were the next most abundantly produced chemokines, respectively (Figure 5B). Activin-A had no effect on the constitutively low amounts (5-10 pg/mL) of MCP-1 produced by NK cells (Figure 5B).

The effects of activin-A on NK-cell cytokine and chemokine production. NK-cell supernatants generated from experiments shown in Figure 4 were screened with the use of (A) a multiplex cytokine and (B) a multiplex chemokine array system and levels measured on a Luminex instrument. The concentration range produced by individual donors is shown in nanograms per milliliter. Data represent the mean plus or minus 1 SD of 3 separate donors. *P < .01 versus no activin-A.
The effects of activin-A on NK-cell cytokine and chemokine production. NK-cell supernatants generated from experiments shown in Figure 4 were screened with the use of (A) a multiplex cytokine and (B) a multiplex chemokine array system and levels measured on a Luminex instrument. The concentration range produced by individual donors is shown in nanograms per milliliter. Data represent the mean plus or minus 1 SD of 3 separate donors. *P < .01 versus no activin-A.
Activin-A suppresses NK-cell proliferation but not lytic function
Cellular proliferation is an important component of NK-cell function. We thus assessed the possible effects of activin-A in this process. To do this, CFSE-labeled NK cells were cultured with cytokines and TLR ligands with or without the addition of recombinant activin-A for 5 days. Under these conditions, a greater percentage of CD56bright NK cells divided compared with their CD56int counterparts but, strikingly, each population's cell division was strongly suppressed in the presence of activin-A (Figure 6 and typical analysis shown in Figure S3).

The effects of activin-A on NK-cell proliferation. Purified, CFSE-labeled NK cells were cultured at 105 in 96-well plates in media supplemented with IL-2 (20 U/mL) + IL-12 (10 ng/mL) with or without addition of poly I:C (12.5 μg/mL) or LPS (100 ng/mL) in the absence or presence of activin-A (100 ng/mL) for 5 days. The level of NK-cell proliferation was assessed through the dilution of CFSE intensity by flow cytometry. Data represent the mean plus or minus 1 SD of 3 separate donors. **P < .05 and *P < .01 versus no activin.
The effects of activin-A on NK-cell proliferation. Purified, CFSE-labeled NK cells were cultured at 105 in 96-well plates in media supplemented with IL-2 (20 U/mL) + IL-12 (10 ng/mL) with or without addition of poly I:C (12.5 μg/mL) or LPS (100 ng/mL) in the absence or presence of activin-A (100 ng/mL) for 5 days. The level of NK-cell proliferation was assessed through the dilution of CFSE intensity by flow cytometry. Data represent the mean plus or minus 1 SD of 3 separate donors. **P < .05 and *P < .01 versus no activin.
Finally, as with IFN-γ production and cellular proliferation, NK-cell cytotoxicity is a critical component of immune surveillance. To test whether activin-A could influence NK-cell cytotoxicity, we stimulated NK cells with either a high dose of IL-2 (100 U/mL; Figure 7A) or a combination of IL-2 (20 U/mL) and IL-12 (Figure 7B) with or without addition of activin-A or TGF-β for 3 days. These cells were then used as effectors in a cytotoxicity assay with the use of calcein-labeled K562 or Jurkat cells as targets. In striking contrast to TGF-β, which significantly suppressed the ability of NK cells to lyse each target cell type, activin-A was unable to suppress NK cell–mediated cytotoxic function under any of the conditions tested (Figure 7A,B). Importantly, activin-A suppressed IFN-γ secretion by NK cells stimulated with high-dose IL-2 as potently as TGF-β and also suppressed IL-2– and IL-12–induced IFN-γ production, albeit less potently than TGF-β, during this extended culture period (Figure 7C). To investigate the mechanism underlying these differences, the same NK-cell populations used in the killing assay shown in Figure 7A, as well as the NK donors used in Figure 7C, were stained for expression of CD69 and the NCRs NKp30 and NKG2D. First, stimulation of NK cells with either high-dose IL-2 or the combination of IL-2 and IL-12 resulted in up-regulation of each of these molecules compared with levels expressed ex vivo (Figure 8A-F). Most importantly, although TGF-β substantially down-regulated the levels of CD69, NKp30, and NKG2D expressed on NK cells, activin-A only weakly down-regulated these cell surface receptors (Figure 8A-F).

The effects of activin-A and TGF-β on NK-cell killing and IFN-γ production. Cytotoxicity of 2 × 104 calcein-labeled K562 or Jurkat cells by purified NK cells cultured at 106 in 24-well plates for 3 days in (A) media supplemented with IL-2 (100 U/mL) or (B) media supplemented with IL-2 (20 U/mL) + IL-12 (10 ng/mL) in the absence or presence of activin-A or TGF-β (each 100 ng/mL). Killing efficiency was determined by calcein dye release and measured by spectrometry. (C) NK-cell supernatants collected after the 3 days of culture were measured for IFN-γ production by ELISA. Data represent the mean plus or minus 1 SD of 3 separate donors. *P < .01 versus other groups.
The effects of activin-A and TGF-β on NK-cell killing and IFN-γ production. Cytotoxicity of 2 × 104 calcein-labeled K562 or Jurkat cells by purified NK cells cultured at 106 in 24-well plates for 3 days in (A) media supplemented with IL-2 (100 U/mL) or (B) media supplemented with IL-2 (20 U/mL) + IL-12 (10 ng/mL) in the absence or presence of activin-A or TGF-β (each 100 ng/mL). Killing efficiency was determined by calcein dye release and measured by spectrometry. (C) NK-cell supernatants collected after the 3 days of culture were measured for IFN-γ production by ELISA. Data represent the mean plus or minus 1 SD of 3 separate donors. *P < .01 versus other groups.

The effects of activin-A and TGF-β on NK-cell CD69, NKp30, and NKG2D expression. NK cells from Figure 7A and 7C were stained with monoclonal antibodies against CD56, CD69, NKp30, and NKG2D. Ex vivo levels are compared with levels expressed after 3 days of culture and measured by flow cytometry. One representative donor of 6 is shown.
The effects of activin-A and TGF-β on NK-cell CD69, NKp30, and NKG2D expression. NK cells from Figure 7A and 7C were stained with monoclonal antibodies against CD56, CD69, NKp30, and NKG2D. Ex vivo levels are compared with levels expressed after 3 days of culture and measured by flow cytometry. One representative donor of 6 is shown.
These results highlight an important biological difference between the actions of activin-A and TGF-β on NK-cell function with activin-A–suppressing NK-cell IFN-γ production but not killing capacity. Taken together, our findings have identified a novel regulatory role for activin-A in NK-cell function that is distinct from TGF-β and is important considering the impact of DC and NK-cell cross-talk in innate and adaptive immune responses.
Discussion
Activin-A was first described as a factor that accentuates the release of FSH15 and is well studied with regard to developmental and reproductive processes.30,31 More recently it has become clear that activin-A plays diverse roles in a variety of biological processes, including erythropoiesis32 and thymopoiesis,33 stem cell differentiation and pluripotency,22,34 neural protection,35 apoptosis,36 and carcinogenesis.16,17 Most recently, we have demonstrated that activated human DCs make large amounts of activin-A12 and our current report demonstrates for the first time a regulatory role of activin-A in NK-cell function.
Injection of LPS or poly I:C into mice results in increased systemic levels of activin-A within hours (D. J. Phillips, personal communication, February 2008). These TLR ligands are known to also be potent inducers of DC and NK-cell activation in vivo because they constitute danger signals sensed by these cells during pathogenic bacterial or viral infection.37,38 At the onset of infection, pathogen-exposed DCs can form epicenters of inflammation and produce inflammatory chemokines such as IL-8, MIP-1α, and IP-10 that induce NK-cell locomotion and recruitment.8,39,40 The ability of these activated DCs to positively influence NK-cell function is well documented and can occur via receptor dependent mechanisms that act in concert with DC-derived Th1-inducing cytokines such as IL-12, IL-15, IL-18, and type I interferons that act as potent cofactors for optimal NK-cell activation.41-44 Interestingly, these same danger signals stimulate DCs to produce large amounts of activin-A,12,45 suggesting the simultaneous induction of a negative feedback loop for minimizing the development of immunopathology during immune induction.
We have previously described an important demarcation in the regulatory properties of DC-derived activin-A in inflammatory processes. When MoDCs are activated with CD40L they produce proinflammatory chemokines such as monokine induced by IFN-gamma (MIG), IL-8, MCP-1, IP-10 and regulated on activation normal T-cell expressed and secreted (RANTES), together with the cytokines IL-6, 10, 12, and TNF-α.12 However, these same DCs simultaneously release large amounts of activin-A12 and if this activin-A is neutralized by the addition of its natural antagonist follistatin, the result is a dramatic increase in the secretion of these proinflammatory mediators by DCs.12 Therefore, in the context of CD40/CD40L stimulation, activin-A acts as an autocrine regulator, attenuating the DCs proinflammatory profile. Interestingly, this activity appears restricted somewhat to CD40L stimulation, because the same effect was not observed when MoDCs were activated with LPS or intact E coli in the presence of follistatin.12
With DC and NK-cell cross-talk now recognized as critical for the generation of protective immunity against tumor or virus-infected cells,4 we sought to identify a novel relationship between activin-A production by DCs and regulation of NK-cell function. Our initial findings that ex vivo–purified NK cells express activin and TGF-β type I and II receptor mRNAs and that activin receptor mRNA expression was further up-regulated in response to specific cytokine stimulation provided the first indication that human NK cells are potentially receptive to activin-A. This finding was confirmed when addition of exogenous recombinant activin-A to NK cells resulted in enhanced Smad 2/3 signaling. Of particular interest are the apparent divergent effects of activin-A and TGF-β in the regulation of activin and TGF-β receptor mRNA expression. The addition of activin-A or TGF-β to IL-2– and IL-12–stimulated NK cells resulted in additional up-regulation of message for the activin type I receptor, ALK4, but simultaneously down-regulated type I and II TGF-β receptor mRNAs. This finding possibly indicates that NK cells exposed to activin-A or TGF-β subsequently become less receptive to regulation mediated by TGF-β but simultaneously more receptive to regulation mediated by activin-A.
The present report indicates that activin-A is as potent as TGF-β, a known suppressor of NK-cell function,46,47 in inhibiting IFN-γ production by activated NK cells and that exposure to activin-A modulates the chemokine and cytokine profile NK cells express. Moreover, activin-A down-regulates CD25 expression and suppresses NK-cell proliferation in response to cytokine and TLR stimulation. This finding is intriguing because we found activated human MoDCs and CD1c+ PBDCs to be potent producers of activin-A yet poor producers of TGF-β.12 Our finding that activated DCs produce large amounts of activin-A (but not TGF-β)12 and that activin-A potently down-regulates NK-cell function provides the first evidence for a novel regulatory mechanism used by DCs in the control of innate immune responses.
This mechanism of regulation may be DC subset dependent. Live E coli, specific TLR ligands, and CD40L induce MoDCs to produce large amounts of activin-A whereas CD1c+ PBDCs spontaneously produce activin-A upon in vitro culture alone.12 Plasmacytoid DCs (pDCs), however, do not produce activin-A in vitro even when stimulated with TLR ligands such as poly I:C, R848, or CpG or influenza virus.12,45 These ligands are known to stimulate pDCs to produce large amounts of type I interferons, which enhance very early activation of NK cells.48,49 Thus, pDCs potentially do not possess this mechanism of NK-cell regulation, and this finding highlights that interactions between pDCs and NK cells may be qualitatively different to those of other DC subsets.
In humans, 2 major subsets of NK cells have been identified and divided according to CD56 expression levels.50 The CD56intermediate population represents 90% to 95% of NK cells circulating in human blood, and these cells possess immediate cytolytic function, whereas CD56bright NK cells preferentially localize in lymph nodes, proliferate in response to stimulation, and secrete high levels of IFN-γ.51 In agreement with these studies, we also found that a greater percentage of CD56bright NK cells produced IFN-γ (data not shown) and proliferated in response to cytokine and TLR ligand stimulation compared with the CD56intermediate population. Most importantly, activin-A effectively suppressed the proliferation and global IFN-γ production of both NK-cell populations.
NK cells also express both killer Ig-like inhibitory receptors (KIRs)52,53 and NCRs such as NKp30 and NKG2D.54 NCR and NCR ligand engagement and the lack of KIR engagement results in NK-cell activation and lysis of cells expressing low levels of self-human leukocyte antigen (HLA)–A, -B, or -C alleles.55 Our finding that activin-A regulates NK cell IFN-γ production and proliferation but only marginally down-regulates NKp30 and NKG2D expression and does not suppress killing function defines a novel regulatory pathway likely engaged during pathogen infection and NK and DC cross-talk.
Regulation of NK cell–derived IFN-γ in the lymph node has important implications for the quality of the emerging primary T-cell immune response. NK cells in the lymph node are thought to provide an early source of IFN-γ that is required for Th-1 polarization of CD4+ T cells.11 Furthermore, during DC and NK-cell cross-talk, NK cell–derived IFN-γ enhances DC activation and results in more efficient antigen processing and cross presentation for antigen-specific CD8+ T-cell priming.56,57 IFN-γ also can positively affect the quality of antigen-specific CD8+ T-cell responses. Antigen-specific CD8+ T cells exposed to IFN-γ during priming expand more robustly, are more efficient killers, and give rise to a better-quality memory repertoire.58,59
Together with activating signals, the immune system often coordinately expresses regulators such as TGF-β, prostaglandin E2 (PGE2), adenosine triphosphate (ATP), or IL-10, which modulate immune cell activation.29,47,60,61 In this way, the ability of activin-A to regulate NK-cell IFN-γ production may be critical in preventing inappropriate immune cell activation, tissue damage, or cytokine-induced shock and mortality. Taken together, our results define a novel regulatory capacity for DC-derived activin-A that encompasses both innate and adaptive immunity.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
We thank Drs David Phillips, Mark Smyth, Jeremy Swan, Suzanne Graham, Oliver Klein, and Lisa Ebert for helpful discussion and Dr Brent McKenzie for additional technical assistance.
N.R. was supported by an Industry Fellowship Grant from the Australian National Health and Medical Research Council (NHMRC, Canberra, Australia) and the Ludwig Institute for Cancer Research (Melbourne, Australia). E.M. is an employee of CSL Limited and an Honorary Senior Research Fellow of the Ludwig Institute for Cancer Research. J.C. is an NHMRC Practitioner Fellow.
Authorship
Contribution: N.R. designed research, performed research, collected and analyzed data, and wrote the paper; H.W., T.M.A., and N.K. performed research and collected and analyzed data; and J.C. and E.M. designed research, analyzed data, and wrote the paper.
Conflict-of-interest disclosure: E.M. is employed by CSL Limited, but no reagent or potential product of CSL was used in this article, nor does this author have any financial interest in this work. All other authors declare no competing financial interests.
Correspondence: Neil Robson, Ludwig Institute for Cancer Research, Melbourne Centre for Clinical Sciences, Austin Health, Heidelberg, Victoria, 3084, Australia; e-mail: neil.robson@ludwig.edu.au.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal