

Abstract
The progress made in the understanding of chronic myeloid leukemia (CML) since the recognition of a common chromosomal abnormality to the introduction of ever more effective tyrosine kinase inhibitors is unprecedented in cancer. The expected survival for patients diagnosed with CML today, if properly managed, is probably similar to that of the general population. When managing patients with CML the goal is to achieve the best long-term outcome and we should base the treatment decisions on the data available. The results from cytogenetic and molecular analyses have to be interpreted judiciously and all available treatment options integrated into the treatment plan properly. The availability of several treatment options in CML is an asset, but the temptation of rapid succession of treatment changes because of perceived suboptimal response or for adverse events that could be managed needs to be avoided. Any decision to change therapy needs to weigh the expected long-term outcome with the current option versus the true expectations with any new option, particularly as it relates to irre-versible outcomes, such as transformation to blast phase and death. In this manuscript, we discuss the treatment approach that has helped us manage successfully a large CML population.
Introduction
Not long ago, 2 main treatment options existed for patients with chronic myeloid leukemia (CML): IFN and stem cell transplantation (SCT). IFN, although effective and even curative in some patients,1 had significant toxicity that limited its universal appeal. SCT had a recognized curative potential but was applicable to few patients and carried significant morbidity and mortality. Patients without adequate response were limited to palliative management or offered investigational options. Tyrosine kinase inhibitors (TKIs) changed our approach. The treatment algorithms have changed, and so have the treatment goals, the monitoring tools, and the expectations of patients and physicians. These changes have been very dynamic. How we perceived TKI therapy 5 years ago is not how we think about it today, and how we see it today is not how it may be in the near future. However, a treatment of relative simplicity, with oral agents and much better tolerance than treatments of years past, requires strict adherence to principles that provide the best hope for a long-term favorable outcome. Many of the basic management principles in CML are not without controversy, with diverging opinions that reflect the constant evolution of our thinking and the need for additional research. Here we will discuss our personal approach to the management of CML, recognizing that some of these approaches may be handled differently by other esteemed CML experts.
Diagnosis, staging, and risk classification
The diagnosis of CML can be done with peripheral blood. An elevated WBC count with left shift, frequently with basopilia, and an enlarged spleen is suggestive of CML. Although the presence of BCR-ABL can be confirmed with FISH or PCR for BCR-ABL, a BM aspiration and cytogenetic analysis is mandatory for proper workup. Without this, one cannot tell if there might be increased blasts or basophils that would shift the staging from chronic (CP) to accelerated (AP) or blastic (BP) phase. In addition, we would not know whether additional chromosomal abnormalities are present besides the Philadelphia chromosome (ie, clonal evolution). If cytogenetics are not done at diagnosis and later identified such changes, it is impossible to determine whether this is a new occurrence or persistence of previously present abnormalities. Thus, in all patients with suspected CML we perform a BM aspiration with differential count and cytogenetics as well as FISH and PCR at diagnosis to confirm that the fusion gene present is detectable. We give no consideration to the absolute value of the transcript levels at diagnosis, but uncommon rearrangements (eg, e13a3, e14a3, e19a2) may not be detectable by the standard PCR probes. Not having a baseline PCR on a patient with such abnormality could create confusion when a subsequent evaluation comes with undetectable transcripts. This could be interpreted as complete molecular response (CMR) when indeed it represents an inevaluable test. Probes are available that can detect these transcripts,2 but they are usually not routinely used in most commercial and academic laboratories.
It is important to stage patients into CP, AP, and BP. There are many classifications, including one by the World Health Organization.3 We prefer and use the stage classification developed in the 1990s (Table 1)4 because it is backed by data. All major studies with TKI have used these definitions. The proposal, for example, to move the threshold for BP from more than or equal to 30% to more than or equal to 20% blasts is not backed by data. Analysis of patients treated with TKI showed that patients with 20% to 29% blasts have an outcome similar to patients with AP criteria, having a median survival approximately 12 months longer than those with blasts more than or equal to 30%.5 Unquestionably, both thresholds are arbitrary, and we would prefer a more biologically relevant classifications that eliminates the arbitrariness of these thresholds (eg, by gene expression profile).6 Some progress has been made in this regard, but gene expression signatures are not widely available and results are variable. It is also difficult to assess subjective criteria, such as persistent thrombocytosis unresponsive to therapy without defining what therapy is meant by this statement. The issue is not only semantic. The use of different criteria results in stage migration, a phenomenon by which, using different criteria, appears to improve the outcome of patients just from reclassification.7 By reclassifying patients with 20% to 29% blasts from AP to BP, we would see better outcomes for both groups without any real improvement. We need to use a classification that helps understand and explain to patients what the expected results are.
Staging of CML that we use in our practice
| Chronic phase |
| None of the criteria for accelerated or blastic phase |
| Accelerated phase |
| Blasts ≥ 15% in blood or BM |
| Blasts plus progranulocytes ≥ 30% in blood or BM |
| Basophilia ≥ 20% in blood or BM |
| Platelets < 100 × 109/L unrelated to therapy |
| Cytogenetic clonal evolution |
| Blast phase |
| ≥ 30% blasts in blood or BM |
| Extramedullary disease with localized immature blasts |
| Chronic phase |
| None of the criteria for accelerated or blastic phase |
| Accelerated phase |
| Blasts ≥ 15% in blood or BM |
| Blasts plus progranulocytes ≥ 30% in blood or BM |
| Basophilia ≥ 20% in blood or BM |
| Platelets < 100 × 109/L unrelated to therapy |
| Cytogenetic clonal evolution |
| Blast phase |
| ≥ 30% blasts in blood or BM |
| Extramedullary disease with localized immature blasts |
For patients in CP, it is common to use one of the available risk stratification scores, such as Sokal,8 Hasford,9 or the more recently developed EUTOS score.10 We prefer to use the Sokal score because it has been more consistently predictive of outcome. In the TKI era, it has been shown to correlate with response to imatinib, with event-free survival (EFS), and even with the probability of maintaining a durable CMR after treatment discontinuation. The EUTOS score has the beauty of simplicity, including only 2 factors: spleen size and basophils. Unfortunately, 2 independent series have not confirmed its value, albeit with some methodologic differences in the analysis.11,12 In our practice, we do not make treatment decisions based on Sokal score, and we offer all patients treatment with TKIs as outlined under “Initial treatments.” The outcome of all groups has improved significantly with TKIs, particularly with the new agents, and there is little evidence that Sokal-specific interventions have any additional value over management based on close follow-up of patients and adhering to the relevant treatment goals.
Initial treatment
When a patient has suspected CML without confirmation, we initiate hydroxyurea if the WBC is elevated (eg, > 80-100 × 109/L), to reduce WBC counts close to normal levels. We continue hydroxyurea until confirmation of the Philadelphia chromosome. If the WBC is very high, allopurinol is used to minimize complications associated with tumor lysis. Once the diagnosis is confirmed, we start therapy with a TKI. It is not necessary to bring the WBC down to normal levels before starting TKIs; and once we start the TKI, hydroxyurea is discontinued.
Our initial therapy for all CML patients in CP is with a TKI. What TKI should be used has evolved in recent years. From 2000 to 2005, we treated all patients with imatinib, and for most of this time we have used high-dose imatinib for all (usually on clinical trials). In our opinion, the results with high-dose imatinib (800 mg daily) are superior to those achieved with standard dose, provided one maintains optimal dose intensity. In our experience, high-dose imatinib results in higher rates of complete cytogenetic response (CCyR) at 36-months (69% vs 58%; on intention-to-treat), major molecular response (MMR; 69% vs 44%), and CMR (55% vs 32%) compared with standard dose.13 More importantly, the rates of transformation and events are lower with high-dose imatinib with 3-year EFS rates of 92% versus 85%, and transformation-free survival (TFS) rates of 97% versus 89%.13 The use of high-dose imatinib, however, is controversial. Two randomized studies, one in high-risk Sokal patients14 and another in all patients,15 showed no long-term benefit for patients treated with high-dose imatinib. The latter study suggested that patients achieved CCyR and MMR considerably earlier with high-dose (6-month CCyR 57% vs 45% with standard dose; P = .0145), but the rates eventually reached the same level (12-month CCyR 70% vs 66%, respectively; P = not significant). Importantly, patients able to maintain adequate dose intensity derived a significant benefit. In contrast, a more recent study suggested higher rates of CCyR and MMR for patients treated with high-dose imatinib, with 12-month rates of MMR of 59% with imatinib 800 mg versus 44% with imatinib 400 mg (P < .001). By 3 years, the rates became equivalent (82% and 79%, respectively), but there was a sustained improvement in the CMR rate (57% with imatinib 800 mg vs 46% with imatinib 400 mg).16 Importantly, there was also a suggestion of an improved EFS with high-dose imatinib. Overall, it is fair to state that, even if there is a benefit with high-dose imatinib, the use of high-dose imatinib is perhaps not applicable to all patients. With better options available, the role of high-dose imatinib might be limited to patients who have no access to the newer TKIs. If high-dose imatinib is to be used, early recognition and adequate management of adverse events (Table 2) are required to optimize tolerability and produce optimal results.
Our approach to management of common adverse events with TKIs in CML
| Adverse events | Management |
|---|---|
| Nonhematologic adverse events | |
| Nausea and vomiting | Take imatinib with food; antiemetics if necessary |
| Diarrhea | Loperamide or diphenoxylate atropine |
| Fluid retention | |
| Peripheral edema | Diuretics as needed (usually furosemide) |
| Periorbital edema | Steroid-containing cream |
| Pleural effusion | Observation if minimal; when intervention is required, stop TKI, use diuretics, corticosteroids may help in occasional patients; resume TKI with dose reduction when the effusion has significantly improved; thoracentesis if effusion not resolving or large and symptomatic |
| Skin rash | Symptomatic therapy (eg, antihistamines); topical steroids; occasionally systemic steroids; minimize sun exposure |
| Muscle cramps | Tonic water or quinine; calcium gluconate may sometimes help; electrolyte replacement if needed (eg, potassium) |
| Arthralgia, bone pain | NSAID (should be used with caution if platelet dysfunction is suspected, eg, with dasatinib) |
| Elevated transaminases | Monitor if grade 1 or 2; interrupt therapy if grade 3; restart a lower dose when recovered to grade ≤ 1; corticosteroids may help some patients if recurrent |
| Elevated bilirubin | Monitor if grade 1 or 2; interrupt therapy if grade 3; restart a lower dose when recovered to grade ≤ 1; elevation of bilirubin common with nilotinib, particularly among patients with Gilbert syndrome; in those instances, may allow continuation of therapy in some instances with grade 3 |
| Elevated lipase, amylase (asymptomatic) | Monitor if grade 1 or 2; interrupt therapy if grade 3; restart at lower dose when recovered to grade ≤ 1 |
| Hyperglycemia | More common with nilotinib; stop therapy if grade ≥ 3; restart therapy when recovered to grade ≤ 1 with reduced dose; no contraindication to use nilotinib in patients with diabetes mellitus; close monitoring and adjustment of hypoglycemic agents as needed |
| Hematologic adverse events | |
| Neutropenia | Hold therapy if grade ≥ 4 (ie, ANC < 0.5 × 109/L);† restart at the same dose if recovery to ANC ≥ 0.75 × 109/L within 2 wks; restart at lower dose if recovery after 2 wks; consider filgrastim if recurrent/persistent, or sepsis72 * |
| Thrombocytopenia | Hold therapy if platelets < 40 × 109/L†; restart at the same dose if recovery within 2 wks to ≥ 75 × 109/L; restart at lower dose if recovery after 2 wks; consider IL-11 10 μg/kg 3-7 d/wk73 * |
| Anemia | Treatment interruption/dose reduction usually not indicated; consider erythropoietin or darbepoetin74 *; transfusions rarely needed |
| Adverse events | Management |
|---|---|
| Nonhematologic adverse events | |
| Nausea and vomiting | Take imatinib with food; antiemetics if necessary |
| Diarrhea | Loperamide or diphenoxylate atropine |
| Fluid retention | |
| Peripheral edema | Diuretics as needed (usually furosemide) |
| Periorbital edema | Steroid-containing cream |
| Pleural effusion | Observation if minimal; when intervention is required, stop TKI, use diuretics, corticosteroids may help in occasional patients; resume TKI with dose reduction when the effusion has significantly improved; thoracentesis if effusion not resolving or large and symptomatic |
| Skin rash | Symptomatic therapy (eg, antihistamines); topical steroids; occasionally systemic steroids; minimize sun exposure |
| Muscle cramps | Tonic water or quinine; calcium gluconate may sometimes help; electrolyte replacement if needed (eg, potassium) |
| Arthralgia, bone pain | NSAID (should be used with caution if platelet dysfunction is suspected, eg, with dasatinib) |
| Elevated transaminases | Monitor if grade 1 or 2; interrupt therapy if grade 3; restart a lower dose when recovered to grade ≤ 1; corticosteroids may help some patients if recurrent |
| Elevated bilirubin | Monitor if grade 1 or 2; interrupt therapy if grade 3; restart a lower dose when recovered to grade ≤ 1; elevation of bilirubin common with nilotinib, particularly among patients with Gilbert syndrome; in those instances, may allow continuation of therapy in some instances with grade 3 |
| Elevated lipase, amylase (asymptomatic) | Monitor if grade 1 or 2; interrupt therapy if grade 3; restart at lower dose when recovered to grade ≤ 1 |
| Hyperglycemia | More common with nilotinib; stop therapy if grade ≥ 3; restart therapy when recovered to grade ≤ 1 with reduced dose; no contraindication to use nilotinib in patients with diabetes mellitus; close monitoring and adjustment of hypoglycemic agents as needed |
| Hematologic adverse events | |
| Neutropenia | Hold therapy if grade ≥ 4 (ie, ANC < 0.5 × 109/L);† restart at the same dose if recovery to ANC ≥ 0.75 × 109/L within 2 wks; restart at lower dose if recovery after 2 wks; consider filgrastim if recurrent/persistent, or sepsis72 * |
| Thrombocytopenia | Hold therapy if platelets < 40 × 109/L†; restart at the same dose if recovery within 2 wks to ≥ 75 × 109/L; restart at lower dose if recovery after 2 wks; consider IL-11 10 μg/kg 3-7 d/wk73 * |
| Anemia | Treatment interruption/dose reduction usually not indicated; consider erythropoietin or darbepoetin74 *; transfusions rarely needed |
NSAID indicates nonsteroidal anti-inflammatory drug; and ANC, absolute neutrophil count.
The use of erythropoietin, darbepoietin, filgrastim, and IL-11 in this setting is not standard and should be considered investigational.
The standard recommendation is to hold if grade ≥ 3 (ie, ANC < 1 × 109/L, platelets < 50 × 109/L).
Since 2005, we have offered dasatinib and nilotinib as initial therapy to all our patients, first in clinical trials and now as standard of care. The phase 2 studies initiated then have shown remarkable rates of early responses, with nearly 90% of patients achieving a CCyR by 3 months of therapy with either agent, and excellent EFS, TFS, and survival.13,17,18 Recent randomized trials have confirmed these excellent results, with higher rates of CCyR and MMR and lower rates of transformation with both dasatinib and nilotinib.19-22 These results show that dasatinib and nilotinib provide better outcome to patients with CP CML. However, it is important to discuss some possible caveats to the universal use of these agents and to analyze the pros and cons of each approach.
Results with nilotinib and dasatinib as frontline therapy in single-institution trials at MD Anderson Cancer Center have shown very high rates of CCyR and MMR, which are superior to those in historical populations treated with standard-dose imatinib on an intention-to-treat analysis.13 The 36-month rates of CCyR are 58% for imatinib, 76% for nilotinib, and 78% for dasatinib. The MMR rates are 44%, 73%, and 76%, respectively. Importantly, the rates of CMR were 32% with imatinib 400 mg, 59% with nilotinib, and 52% with dasatinib. Rates of EFS were significantly higher with dasatinib or nilotinib (91%-95%) than with standard-dose imatinib (85%), with similar trends for TFS (97%-100% vs 89%, respectively). Interestingly, the 3-year rates of EFS (92%) and TFS (97%) with high-dose imatinib appear similar to those with nilotinib and dasatinib.13 Randomized trials have confirmed the results with second-generation TKI (2G-TKI). The ENESTnd study, comparing nilotinib versus imatinib, has shown a cumulative rate of MMR by 3 years of 70% to 73% with nilotinib and 53% with imatinib, and a significantly lower rate of transformation to AP or BP (2.1%-3.2% vs 6.7%, respectively).21,22 The DASISION study also reported a benefit for dasatinib-treated patients with cumulative MMR rates by 24 months of 64% with dasatinib and 46% with imatinib, and rates of transformation to AP or BP of 3.5% and 5.8%, respectively.19,20
The use of IFN in combination with imatinib has been suggested in 2 randomized trials to improve the rate of MMR23 or superior molecular response.24 However, our own randomized study25 as well as the CML IV study16 did not show any benefit in response rate (cytogenetic or molecular) for patients treated with the combination compared with imatinib alone, and none of the studies showed an improvement in EFS or overall survival. In addition, the addition of IFN adds toxicity and cost to the therapy. Thus, we do not use this combination in any patient.
The data thus suggests better outcomes with 2G-TKI as initial therapy for CML. However, several aspects are not addressed by the mere enumeration of the results of these studies. One important deficiency of these studies is that the benefit of each drug is considered in isolation, not accounting for the effect of subsequent therapy. In CML, we are fortunate to have effective therapy for patients who experience failure to initial therapy (more on this under “When do we change therapy?”). In addition, most patients who experience such failure are generally in good condition and not at immediate risk of dying. Thus, salvage therapy can adequately rescue many patients who do not have optimal response to initial treatment. The studies, as they have been designed today, address only the issue of what drug is better (eg, imatinib vs the new TKIs). This approach ignores our ability to rescue patients with effective second-line therapies that may allow for prolonged EFS (ie, considers an event as an irreversible occurrence). The real question is what strategy is better: the use of second-generation drugs as initial therapy or after failure to imatinib. These studies have not been done, but the effect of subsequent therapy can be accounted for by measuring what has been called current EFS. In such an analysis, a patient who, for example, loses a CCyR counts as an event. If that patient then responds to second-line therapy, the event is reversed. With such an approach, the 7-year EFS with imatinib is 81%, but the current EFS is 88%.26 This is a more accurate estimate of what is achieved with the sequential use of TKI.
The counterargument is that with imatinib, based on results of the IRIS study27 and from intention-to-treat or real-life reports,28,29 only approximately two-thirds of patients have an acceptable outcome if we consider CCyR as the minimum acceptable response, and considering as failure to therapy discontinuation of therapy for toxicity or other reasons. Among those who do not do well on imatinib, only approximately 50% achieve a CCyR with 2G-TKI, and 10% to 15% of them have lost their response.30,31 Although the data with 2G-TKI used as frontline therapy is still maturing, the rate of CCyR is significantly improved, with rates of more than 90% and very low rates of transformation in the first 2 to 3 years, the years with the greatest risk of transformation. Undoubtedly, some patients will eventually lose their response, but it is also clear that some of these patients may be rescued with new agents, such as ponatinib.32,33 Thus, based on these extrapolations, we estimate that more patients will ultimately have a favorable outcome when treated with dasatinib or nilotinib as frontline therapy than with imatinib followed by dasatinib or nilotinib upon failure (Figure 1).

Graphic representation of the estimated outcome with 2 different strategies for frontline CML therapy. (A) Standard-dose imatinib first, followed by salvage therapy with second-generation TKI on failure. (B) Second-generation TKI first.
Graphic representation of the estimated outcome with 2 different strategies for frontline CML therapy. (A) Standard-dose imatinib first, followed by salvage therapy with second-generation TKI on failure. (B) Second-generation TKI first.
Thus, we come back to our position that we recommend 2G-TKI as initial therapy for all patients because we think that, in the long term, most patients will do better with this strategy. However, we recognize that many patients can be treated well with imatinib. Perhaps two-thirds of all CML patients may do well with imatinib (more with high-dose) provided they receive optimal management, including maintaining optimal dose intensity and adequate monitoring. It would be useful to define parameters that may identify patients likely to do well with imatinib. Sokal risk score is advocated as such a parameter, but we need better prognosticators, as even some patients with high-risk Sokal do well on imatinib, and many with low-risk scores still benefit from 2G-TKI over imatinib. In any event, if a patient is to be treated with imatinib (eg, because of lack of availability of other agents or because of cost), we would chose high-dose imatinib, and I would closely monitor to identify those with less than optimal response at early time points, to consider early treatment changes before true failure emerges. The presence of other warning factors has also been proposed to recommend therapy with 2G-TKI, such as the presence of major route additional chromosomal abnormalities. Some series have reported a poor prognosis for these patients,34 although in our experience patients with additional chromosomal abnormalities have a similar prognosis as those without them.35,36 Similarly, the few patients who present with p190 (e1a2 rearrangement) have a poor prognosis with imatinib.37 Because there are no data to suggest that the outcome is better for patients with additional chromosomal abnormalities or p190 with 2G-TKI, we do not use these features to decide on therapy, although these patients need to be monitored closely.
The patient with CCyR
One common question is what to do with a patient in CCyR but not in MMR or still PCR-positive. Central to this question is what the goal of therapy is. We consider CCyR the gold standard for a good response. The reason is that achieving CCyR is associated with improved survival.1 With better drugs and enhanced monitoring tools, there is interest in achieving deeper responses. MMR has been introduced as a therapeutic goal based on its possible long-term benefit. The data show that, among patients who achieve CCyR, those who also achieve MMR by 18 months have a modest but statistically significant improvement in the probability of 7-year EFS compared with those with CCyR but no MMR (95% vs 86%, respectively).38 But there is no significant difference in TFS or in overall survival. There is the suggestion that patients with MMR have more durable CCyRs, with 7-year estimates of remaining in CCyR of 97% for those with MMR at 18 months versus 74% for those with CCyR but no MMR.38 However, patients who achieved CCyR on imatinib and eventually lose it have the best probability of achieving a subsequent CCyR with 2G-TKI,39,40 with CCyR rates of 84% and EFS at 2.5 years of 93%.40 This means that patients in CCyR but no MMR and who lose CCyR will probably respond to subsequent therapy. This explains why there is no difference in survival or transformation. Achieving a CMR has not been proven to confer any long-term benefit to patients other than the possibility of considering treatment discontinuation. Treatment discontinuation should not be recommended outside of a clinical trial. Based on the data, we consider molecular responses a measure of success, not a measure of failure. This means that, although we prefer (and patients feel more comfortable with) the lowest possible transcript levels and although MMR may offer modest long-term benefit to patients (eg, lower risk of loss of CCyR, slightly higher EFS), not achieving these levels of molecular response does not constitute treatment failure in a patient with CCyR.
These considerations guide our approach to the patient who has achieved CCyR but not MMR, or the patient who has lost MMR but remains in CCyR. In these instances, we do not recommend any change in therapy. We do review with the patient adherence to therapy as this is a common cause of fluctuations in PCR levels, and we may monitor more frequently (eg, every 3 months). But unless the patient loses CCyR, we do not change therapy. This scenario is considered by the European LeukemiaNet recommendations as a suboptimal response, and the treatment recommendations for suboptimal response include continuing therapy unchanged, increasing dose of imatinib, or changing to a different agent.41 This vagueness underscores the lack of data to suggest that any strategy results are better than continuing therapy unchanged. Increasing the imatinib dose or changing to a 2G-TKI may result in lower transcript levels, but it has never been shown that such a treatment change improves the long-term outcome compared with changing therapy at the time of loss of CCyR if this were to happen. In our experience, dose escalation because of increasing transcript levels did not improve the overall or progression-free survival compared with patients in whom the dose was not changed.42
When do we change therapy?
As stated under “The patient with CCyR,” we do not change therapy for changes in transcript levels among patients in CCyR. However, we follow our patients closely to identify true indications of treatment failure that warrant treatment change. Clearly, a patient who has lost a CCyR, or obviously a complete hematologic response, (ie, secondary resistance), needs a different therapy (Table 3). For a patient who has lost CCyR, we change therapy immediately, unless there is reason to believe the CCyR loss is the result of lack of adherence. Delaying treatment change in a patient who has already lost cytogenetic response causes a decreased probability of response and a worse EFS.43 Such delays should be avoided. Our approach to a patient who has primary resistance (ie, who has not achieved predefined treatment goals) is modulated by several factors. The European LeukemiaNet defined treatment failure and suboptimal response to contrast them with what is considered an optimal response. The implications are that patients who meet the definitions of failure should change their treatment. These definitions are evolving and guide only partially what we do clinically (Table 3). Frontline treatment with 2G-TKI has increased our expectations for what we consider optimal response. Deep early responses have been identified for many years to confer a favorable long-term outcome. Patients with transcript levels that decrease by 1-log44 or that are less than 10%45,46 3 months after the start of therapy, or who have not achieved a partial cytogenetic response (PCyR; grossly equivalent to this transcript level),47 have a poorer long-term outcome. Hence, the interest in treating patients with what offers them the deepest and fastest responses.
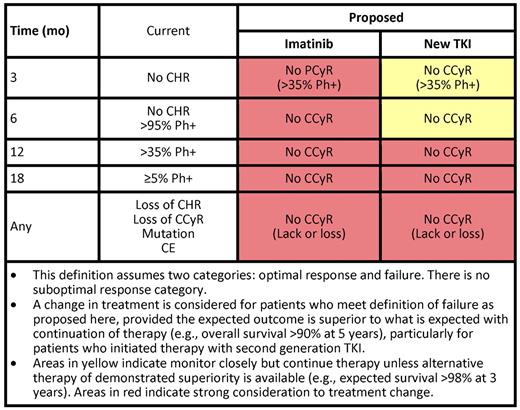
Another important component of our decision to change therapy or not are the risks and benefits expected with the alternative therapy. For example, a patient with CCyR but no MMR after 18 months of therapy has, based on IRIS data, a 96% 7-year probability of being alive without transformation to AP or BP and 96% of being alive.38 If we plan to change therapy, we would seek results that are likely to be better than these expectations. In this setting, we are not aware of such better therapies. As the notion of the favorable long-term prognostic implications of early deeper responses is finally being universally accepted, one interesting scenario is a patient who has not achieved a PCyR (or transcript levels < 10%) after 3 months. Our approach to this patient depends in part on what they have received. If this is a patient treated with imatinib, we would consider changing to a 2G-TKI (Table 3), with the caveat that the long-term benefits of such a change have not been demonstrated. The TIDEL48,49 studies have investigated this early-switch approach and reported positive results by either escalating the dose of imatinib or switching to nilotinib if early goals are not met. These strategies are appealing but need to be confirmed against a control arm where patients maintain their initial therapy unchanged until failure, to confirm that early treatment change improves long-term outcome. For patients receiving a 2G-TKI as frontline therapy, the EFS for those achieving PCyR or less as early as 3 months are less favorable than if they achieved CCyR.50 However, the risk of transformation and death is still minimal. There is no good alternative therapy today that can match an expected survival greater than 98%. Therefore, we would monitor such patients closely but would not offer treatment changes at these early time points. We do consider patients who have not achieved CCyR at later time points (eg, 12-18 months) for treatment change.
Regarding changes in therapy, we are fortunate to have many good options. Both dasatinib30 and nilotinib31 are available for patients who have experienced failure or intolerance to imatinib; other drugs will hopefully soon become available, including bosutinib,51 ponatinib,32,33 and omacetaxine.52 All are valuable agents that have offered clear benefit to many of our patients. Our choice of a second TKI is based on the presence of mutations (which helps only in ∼ 20% of our patients) and the presence of comorbidities that may help select one drug over another. Importantly, in most instances, we have not found the presence of comorbidities to be a contraindication for specific agents. For example, if we need to use nilotinib in a diabetic patient or dasatinib in a patient with history of pulmonary problems, we usually can. We comanage with specialists in the corresponding fields if needed to help the patient tolerate the therapy. Regardless, we are still not satisfied with our available therapies; with dasatinib or nilotinib, only approximately 50% of patients achieve CCyR. This should be improved.
What to do with a patient with a suboptimal response? This category represents a transient and heterogeneous state.53 The significance of a suboptimal response at 6 months (ie, no major cytogenetic response) is very different from one at 18 months (ie, no major molecular response), with the former conferring a prognosis more like that of patients with failure, whereas the latter is more similar to that of an optimal response. The current recommendations for patients with suboptimal response are not clinically helpful as they include all possible options (continue unchanged, increase the dose, or change therapy). In addition, with new agents these criteria are changing as suboptimal response with the established definitions is nearly nonexistent.50 As mentioned earlier, for patients treated with imatinib without a PCyR at 3 months (and definitely at 6 months), we would consider treatment change. By 6 months, we would consider changing therapy to anyone not in CCyR. If the patient is being treated with a 2G-TKI and has not achieved a PCyR despite adequate therapy (ie, without frequent treatment interruptions or intolerance), we may give this patient an additional 3 to 6 months of therapy to try to accomplish the response and monitor closely considering there may not be better treatment alternatives. SCT is frequently mentioned and should be considered; but aside from the risks associated with it and the expected more than 95% survival at 3 years with continued TKI therapy, results of SCT in this setting are unavailable. Fortunately, these scenarios are rare with 2G-TKIs as initial therapy: in our experience, only 1% to 2% of patients meet criteria for suboptimal response or failure at 3 to 12 months. At 18 months, the rate of suboptimal response is higher (12%), but this is a scenario (CCyR with no MMR) where, as discussed, we do not change therapy. In summary, we classify the response of patients into only 2 categories: those in whom we would change therapy (based on a high risk of mortality or transformation) and those in whom we continue therapy unchanged.
A few monitoring pearls
After treatment is started, we do PCR every 3 months for the first 12 months and a cytogenetic analysis every 3 to 6 months. There are reasons why a cytogenetic analysis is important. First, the time to achievement of cytogenetic response has important prognostic implications. Cytogenetic response could be evaluated by FISH that correlates well with cytogenetics and is done on peripheral blood samples. However, we find it important to learn whether a patient is developing chromosomal abnormalities in Ph-negative metaphases, which occur in 10% to 15% of patients.54 A counterargument is that these abnormalities do not represent a reason for treatment change. However, on occasion, patients develop myelodysplastic syndrome or acute leukemia where the malignant clone carries these abnormalities.55 Thus, our failure to understand the significance of this phenomenon is not a reason to ignore it. We also want to identify clonal evolution when it occurs, making the assessment of a karyotype important in patients not in CCyR. Once the patient has achieved a stable CCyR, I perform BM aspirations and cytogenetic analyses much less frequently, particularly relevant among those without additional abnormalities, as these usually develop early during the course of therapy. For these patients, we follow a PCR in peripheral blood every 6 months indefinitely. Monitoring more frequent than every 3 months until CCyR and more frequently than every 6 months after that has little clinical value and may be confusing. When we observe significant transcript increases (ie, ≥ 1-log increase, particularly in patients not in MMR42,56 ), besides assessing compliance, we check PCR again in 1 to 3 months. If the trend is confirmed, we check the BM and cytogenetics.
We only do mutation analysis on patients with resistance, particularly secondary resistance (ie, patients who have lost a hematologic or cytogenetic response). Performing mutation analysis in patients at the time of diagnosis is not recommended, and assessing mutations with suboptimal response or with an increase in transcript levels that does not meet criteria for treatment failure has minimal yield (< 10%).57 We use direct sequencing when doing mutation analysis. This is a technique with low sensitivity, and performing mutations analysis with more sensitive techniques may identify mutations earlier in some patients. Mutations identified by more sensitive methods (eg, multiplexed mass spectrometry assay) might identify mutations at earlier times, and these low-level mutations might be predictive of future outcome.58 However, these methods are not broadly available, and it remains to be proven that intervention when a mutation is detected by these sensitive techniques improves outcome compared with intervention at the time of failure by standard criteria.
Treatment discontinuation
This is a topic of great interest in recent years. There are clear potential benefits derived from permanently discontinuing therapy, not the least of which are the finances of the endless therapy using an expensive drug. However, it is premature to recommend treatment discontinuation in practice. The available data59,60 should give us pause in recommending this broadly. Although the follow-up extends now beyond 2 years, this is still a short follow-up time. Patients who receive allogeneic SCT may relapse more than 5 years after transplantation despite being in continuous CMR, and many times these relapses are in blast phase.61 Thus, the current follow-up is inadequate to properly asses the true risk of treatment discontinuation. Although most relapses have been reported to occur within the first 6 months, some have been reported 19 to 22 months after treatment discontinuation, and not all patients who resume therapy regain CMR.62 There are also questions about the proper selection of patients best suited for treatment discontinuation, including the myriad of questions about the definition of CMR. Many of my patients are interested in learning about this possibility, but most still prefer to continue treatment. We therefore discuss current knowledge about treatment discontinuation with our patients. The few who are interested in stopping, we follow them with PCR every 1 to 2 months for 12 months and then every 2 to 3 months for at least 2 years. This option applies to only patients who have confirmed undetectable BCR-ABL transcript levels for at least 2 years. Our research now focuses on ways to increase the proportion of patients who have undetectable transcripts. Clinical trials are critical to make this option a safe reality for a majority of patients with CML.
Pregnancy
As we aim to return patients to their normal life with successful therapy, a common scenario we face is the issue of pregnancy. There are different ways we encounter this clinical dilemma. One is the patient who wants to become pregnant. For a male patient, there is no formal contraindication for fathering a baby while on TKIs, and the data available suggest that in most instances babies born to such patients have no known abnormalities that can be attributed to TKI.63-65 Still, we advise patients that the information in this setting is limited. For female patients, the issue is much more complex. For a patient who wants to become pregnant, our approach is to advice to plan the pregnancy and try to aim for a response as deep as possible, ideally at least an MMR. Then we interrupt therapy, with a preference for a 3-month washout before conception and for the duration of the pregnancy. We then resume therapy immediately after birth. Patients are followed closely with PCR and FISH in peripheral blood during the pregnancy. When done this way, we have not required restarting therapy in any patient. In some instances, transcript levels may increase; but as long as the patient maintains some cytogenetic response, we do not restart therapy. A more complicated issue is the unplanned pregnancy or the patient diagnosed while pregnant. Although there are anecdotal reports of patients who continue therapy throughout pregnancy with no problems for the baby, we do not recommend using TKI at all during pregnancy. Malformations that are associated with imatinib have been reported when imatinib is continued.64 Occasionally, patients can be followed throughout pregnancy without any intervention.66 For those who need intervention because of a very high WBC or evidence of progression, we prefer to use leukapheresis. Use of hydroxyurea67-69 and IFN69-71 have been reported in pregnancy with no complications, but the experience is limited. We prefer to use short pulses of hydroxyurea to control excessive rises in WBC; IFN can be used, but it may take longer time to obtain a hematologic response and is also associated with more adverse events. Still, much of the current management of CML during pregnancy is mostly empirical and we counsel patients extensively about the risks involved.
In conclusion, most of my patients with CML are doing well with TKI therapy, but we aim to have all patients to do well. For this, we always plan our management approach based on what may offer the best long-term benefit for each patient. We consider it critical to always offer clinical trials that provide optimal care and help answer the remaining questions that will lead to the eventual cure of CML.
Authorship
Contribution: J.C. wrote and reviewed the manuscript and approved the final version; and H.K. reviewed the manuscript and approved the final version.
Conflict-of-interest disclosure: J.C. has received grant support from Novartis, BMS, Pfizer, Ariad, Chemgenex, and Deciphera, and has been a consultant for Novartis, BMS, Pfizer, Ariad, and Chemgenex. H.K. received research support from Novartis, BMS, Pfizer, and Chemgenex.
Correspondence: Jorge Cortes, D. B. Lane Cancer Research, CML & AML Sections, Department of Leukemia, MD Anderson Cancer Center, 1515 Holcombe Blvd, Unit 428, Houston, TX 77030; e-mail: jcortes@mdanderson.org.
