

Key Points
The myeloma transplant B-cell immunome is predictive for response to treatment.
It may be exploited by immunosequencing and library technology as a source for unique target structures and antibodies for immunotherapy.
Abstract
Multiple myeloma (MM) is a hematological cancer for which immune-based treatments are currently in development. Many of these rely on the identification of highly disease-specific, strongly and stably expressed antigens. Here, we profiled the myeloma B-cell immunome both to explore its predictive role in the context of autologous and allogeneic hematopoietic stem cell transplantation (HSCT) and to identify novel immunotherapeutic targets. We used random peptide phage display, reverse immunization, and next-generation sequencing–assisted antibody phage display to establish a highly myeloma-specific epitope fingerprint targeted by B-cell responses of 18 patients in clinical remission. We found that allogeneic HSCT more efficiently allowed production of myeloma-specific antibodies compared with autologous HSCT and that a highly reactive epitope recognition signature correlated with superior response to treatment. Next, we performed myeloma cell surface screenings of phage-displayed patient transplant immunomes. Although some of the screenings yielded clear-cut surface binders, the majority of screenings did not, suggesting that many of the targeted antigens may in fact not be accessible to the B-cell immune system in untreated myeloma cells. This fit well with the identification of heat-shock proteins as a class of antigens that showed overall the broadest reactivity with myeloma patient sera after allogeneic HSCT and that may be significantly translocated to the cell surface upon treatment as a result of immunogenic cell death. Our data reveal a disease-specific epitope signature of MM that is predictive for response to treatment. Mining of transplant immunomes for strong myeloma surface binders may open up avenues for myeloma immunotherapy.
Introduction
Multiple myeloma (MM) is a common hematological malignancy suitable for immunotherapeutic interventions.1 It is characterized by the clonal expansion of malignant paraprotein-producing plasma cells that reside in the highly active immune environment of the bone marrow.2 Myeloma is currently considered incurable for the majority of patients, with allogeneic hematopoietic stem cell transplantation (HSCT) being to date the only treatment modality that reports long-term remissions in a small subset of patients.3-8 However, the majority of myeloma patients are ineligible for this radical form of immunotherapy due to age or medical restrictions. Passive immunotherapy with monoclonal antibodies targeting myeloma cell surface molecules has been preclinically developed over the past decade for some targets such as CD38 or SLAM-F7.9-12 It has shown promise in terms of efficacy and tolerability in advanced clinical trials and may therefore be used in a substantial proportion of patients upon licensing.13-15 Other immunotherapeutic options such as chimeric antigen receptor T cells16-18 or bispecific T-cell engagers19,20 demonstrate encouraging preclinical activity and phase 1 trials are ongoing.21 All of these approaches critically rely on highly and stably expressed myeloma-specific cell surface targets. Compared with monoclonal antibodies, engineered T cells or T-cell–engaging antibodies generally seem to require more tumor-specific expression profiles to ensure a therapeutic index with acceptable safety profile.22 In myeloma, this would imply at least specificity for the B-cell lineage, preferably even complete restriction to the neoplastic cell itself. Tumor (neo-)epitopes are therefore promising therapeutic targets. They are either encoded by the tumor mutanome, harbor tumor-specific posttranslational modifications, or are simply part of tightly regulated or immunologically inaccessible antigens reexpressed in the tumor.23-26 For accessibility reasons the myeloma cell surface represents an important source for therapeutically relevant tumor epitopes. However, identification of potentially targetable epitopes may be cumbersome because they may not easily be detected by gene expression profiling or genome sequencing as these methodologies do not allow for prediction of their posttranslational modifications or surface exposure. An alternative approach to identify such epitopes implies exploitation of B-cell immune responses directed against myeloma-specific antigenic structures. In a previous study, we investigated the antigen specificity of oligoclonal serum antibodies, which transiently emerge in an important percentage of patients after novel agents therapy, autologous or allogeneic HSCT.27-29 These antibodies have been found to confer a good prognosis30-34 and our data suggested that the restricted repertoires contain antibodies recognizing recurrent myeloma antigens.35
Building on these previous data, we now set out to establish an epitope fingerprint of MM by dissecting the transplant B-cell immunome of 18 patients with oligoclonal antibodies to the epitope level. Thereby we established that different patients show largely overlapping epitope recognition profiles that are highly disease-specific and correlate with superior clinical response to treatment with some of the target epitopes being exposed as a result of treatment-induced immunogenic cell death. Moreover, we go on to show how next-generation sequencing (NGS)-assisted screening of antibody repertoires of such patients may serve as an attractive technical tool to identify potentially targetable cell surface structures. Clinically, this discovery platform may support a wide spectrum of immunotherapeutic approaches relevant to the future treatment of myeloma.
Material and methods
Patients and sample characterization
Serum samples of 18 myeloma patients with documented oligoclonal serum antibodies on immunofixation were collected during routine clinical visits after therapy (Table 1). Peripheral blood mononuclear cells (PBMCs) of 3 patients, bone marrow mononuclear cells of 1 patient, and healthy donor PBMCs were used for the construction of phage-displayed antibody libraries. Control serum was obtained from 11 healthy donors, 11 patients after allogeneic or autologous HSCT with malignancies other than myeloma, 6 myeloma patients at first diagnosis, 6 myeloma patients after bortezomib induction, and 6 myeloma patients after allogeneic HSCT without oligoclonal serum antibodies. All patients and healthy donors consented to the use of their biological material for this investigation.
Patient characteristics
| Patient code | Sex | Age, y | Paraprotein isotype | Type of treatment | Time between treatment and sample collection, mo | Response at day 100/12 mo after treatment | Duration of CR at time point of sample collection, mo | GVHD at time point of sample collection | Immuno-suppression at time point of sample collection | Epitope polyreactivity | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| MM031 | M | 50 | IgG λ | Allo-HSCT | 9 | CR/CR | 6 | No | No | 16 | ≥3* |
| MM043 | M | 54 | IgG κ | Allo-HSCT | 7 | CR/CR | 7 | No | CSA | 12 | |
| MM046 | F | 42 | IgA λ | Allo-HSCT | 18 | CR/CR | 15 | Skin | CSA | 9 | |
| MM041 | F | 41 | λ LC | Allo-HSCT | 16 | PR/CR | 12 | No | No | 6 | |
| MM025 | M | 65 | IgG κ | Allo-HSCT | 18 | PR/CR | 18 | No | No | 4 | |
| MM040 | M | 48 | IgG κ | Allo-HSCT | 18 | CR/CR | 20 | No | No | 4 | |
| MM054 | F | 61 | IgG κ | Allo-HSCT | 5 | CR/n.e. | 7 | No | CSA | 4 | |
| MM023 | M | 62 | IgG κ | Allo-HSCT | 5 | CR/CR | 1 | n.e. | n.e. | 3 | |
| MM045 | M | 55 | IgG λ | Allo-HSCT | 14 | CR/CR | 14 | No | No | 2 | <3* |
| MM047 | M | 51 | IgG λ | Allo-HSCT | 14 | vgPR/vgPR | — | No | No | 2 | |
| MM035 | M | 64 | IgA κ | Auto-HSCT | 14 | PR/PD | — | — | — | 2 | |
| MM077 | F | 57 | IgG κ | Auto-HSCT | 3 | vgPR/vgPR | — | — | — | 2 | |
| MM051 | M | 50 | IgG κ | Allo-HSCT | 18 | PR/PD | — | No | No | 1 | |
| MM064 | M | 65 | IgG κ | Auto-HSCT | 7 | PR/PR | — | — | — | 0 | |
| MM073 | M | 43 | κ LC | Bortezomib† | 3 | CR/CR | 4 | — | — | 0 | |
| MM074 | M | 61 | κ LC | Auto-HSCT | 28 | CR/PD | 25 | — | — | 0 | |
| MM075 | M | 73 | IgG λ | Auto-HSCT | 6 | CR/CR | 2 | — | — | 0 | |
| MM076 | F | 72 | κ LC | Auto-HSCT | 7 | CR/CR | 5 | — | — | 0 | |
| Patient code | Sex | Age, y | Paraprotein isotype | Type of treatment | Time between treatment and sample collection, mo | Response at day 100/12 mo after treatment | Duration of CR at time point of sample collection, mo | GVHD at time point of sample collection | Immuno-suppression at time point of sample collection | Epitope polyreactivity | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| MM031 | M | 50 | IgG λ | Allo-HSCT | 9 | CR/CR | 6 | No | No | 16 | ≥3* |
| MM043 | M | 54 | IgG κ | Allo-HSCT | 7 | CR/CR | 7 | No | CSA | 12 | |
| MM046 | F | 42 | IgA λ | Allo-HSCT | 18 | CR/CR | 15 | Skin | CSA | 9 | |
| MM041 | F | 41 | λ LC | Allo-HSCT | 16 | PR/CR | 12 | No | No | 6 | |
| MM025 | M | 65 | IgG κ | Allo-HSCT | 18 | PR/CR | 18 | No | No | 4 | |
| MM040 | M | 48 | IgG κ | Allo-HSCT | 18 | CR/CR | 20 | No | No | 4 | |
| MM054 | F | 61 | IgG κ | Allo-HSCT | 5 | CR/n.e. | 7 | No | CSA | 4 | |
| MM023 | M | 62 | IgG κ | Allo-HSCT | 5 | CR/CR | 1 | n.e. | n.e. | 3 | |
| MM045 | M | 55 | IgG λ | Allo-HSCT | 14 | CR/CR | 14 | No | No | 2 | <3* |
| MM047 | M | 51 | IgG λ | Allo-HSCT | 14 | vgPR/vgPR | — | No | No | 2 | |
| MM035 | M | 64 | IgA κ | Auto-HSCT | 14 | PR/PD | — | — | — | 2 | |
| MM077 | F | 57 | IgG κ | Auto-HSCT | 3 | vgPR/vgPR | — | — | — | 2 | |
| MM051 | M | 50 | IgG κ | Allo-HSCT | 18 | PR/PD | — | No | No | 1 | |
| MM064 | M | 65 | IgG κ | Auto-HSCT | 7 | PR/PR | — | — | — | 0 | |
| MM073 | M | 43 | κ LC | Bortezomib† | 3 | CR/CR | 4 | — | — | 0 | |
| MM074 | M | 61 | κ LC | Auto-HSCT | 28 | CR/PD | 25 | — | — | 0 | |
| MM075 | M | 73 | IgG λ | Auto-HSCT | 6 | CR/CR | 2 | — | — | 0 | |
| MM076 | F | 72 | κ LC | Auto-HSCT | 7 | CR/CR | 5 | — | — | 0 | |
Allo-HSCT, allogeneic HSCT; Auto-HSCT, autologous HSCT; CR, complete remission; CSA, cyclosporine; F, female; GVHD, graft-versus-host disease; LC, light chain; M, male; n.e., not evaluable; PD, progressive disease; PR, partial response; vgPR, very good partial response.
Epitope reactivity and patients' responses revealed a significant association (chi-square test, P = .007). The χ2 testing was performed for time point 12 mo after HSCT.
Bortezomib-based therapy.
Cell lines
All myeloma cell lines (Amo-1, RPMI8226, EJM, U266, KMS-12-BM, IM-9, and LP-1) and control cell lines (KG-1, HL60, Jurkat, and Colo 201) were obtained from American Type Culture Collection (Manassas, VA) or from the German Collection of Microorganisms and Cell Cultures (DSMZ, Braunschweig, Germany) and were cultivated in RPMI 1640 medium (Gibco/Life Technologies, Carlsbad, CA) containing 10% fetal bovine serum (Sigma-Aldrich, St. Louis, MO) and 1% penicillin-streptomycin (Sigma-Aldrich). Cell lines were authenticated by flow cytometry or using Multiplex Cell Authentication (Multiplexion, Heidelberg, Germany) as described recently.36
Random peptide phage display library screening on serum antibodies
Serum antibodies were purified by protein A sepharose chromatography (GE Healthcare, Buckinghamshire, United Kingdom) followed by positive or negative selection on KappaSelect affinity medium (GE Healthcare) to separate κ from λ immunoglobulin fractions as described in the respective product manuals. The linear 12-mer random peptide library was purchased from New England Biolabs (Frankfurt, Germany). Screenings were performed after twofold negative selection on polyclonal immunoglobulin G (IgG; Octapharma, Lachen, Switzerland), random clones were amplified after 3 selection rounds and tested for selective binding to the respective antibody fraction (vs IgG) by enzyme-linked immunosorbent assay (ELISA) as suggested by the manufacturer. Selectively binding phage were sequenced (GATC, Constance, Germany). Phage displaying the random peptide YMTPPLSSQQKS were used as control. All phage were tested for cross-reactivity on all patient serum antibody fractions and controls. Heat-map plotting of this binding study was performed using MATLAB programming language (version R2013b; Mathworks, Natick, MA). Correlation of epitope reactivity with response to treatment was evaluated by χ2 testing.
Recombinant expression of phage-displayed peptide and blocking assay
The oligonucleotide encoding the phage-derived peptide THMWVWDVSPEL was amplified from phage DNA by polymerase chain reaction (PCR), digested with BamHI and EcoRI, ligated into pGEX-2TK (GE Healthcare) for expression as glutathione-S-transferase (GST)-fusion protein in Escherichia coli ER2655 (Invitrogen, Camarillo, CA) and used for a phage competition assay as described previously.37
Parental antigen identification by reverse immunization
Rats were subcutaneously immunized with 1 × 1012 phage particles displaying the peptide THMWVWDVSPEL. Rat sera were screened for the presence of antibodies reactive with the recombinant peptide THMWVWDVSPEL (expressed as GST-fusion protein) by ELISA and western blot. THMWVWDVSPEL-reactive rat serum was subsequently used to immunodetect the parental antigen from the myeloma cell line IM-9 using the serological proteome analysis (SERPA) technology. To this end, a protein extract was generated from the cell line, subjected to 2-dimensional (2D) gel electrophoresis followed by Coomassie brilliant blue staining or western blotting using an anti-rat IgG horseradish peroxidase antibody (Jackson ImmunoResearch, Bar Harbor, ME), essentially as described.35 2D western blots were overlain with the Coomassie-stained reference map by using Delta2D software (version 3.6; Decodon, Greifswald, Germany) and the matching spot was excised for protein identification by mass spectrometry.
scFv bacteriophage library screenings (antibody libraries)
Mononuclear cells were isolated from patient or healthy donor derived blood (for construction of transplant immunome library 1-3 and healthy donor library 1) or bone marrow (for construction of transplant immunome library 4). These single-chain variable fragment (scFv) gene libraries were constructed from the lymphocyte fraction38 using the phage display vector pHAL14 described previously.39 The antibody libraries were packaged with M13KO7 helper phage (NEB). Library selections were performed on whole cells. To eliminate unspecific binders, 1 × 1012 phages were depleted 5 times with 5 × 107 buffy coat cells from healthy donors. Unbound phages were then incubated with 2.5 × 106 target cells (myeloma cell lines or CD71+ cell line) and 5 × 107 control cells (healthy PBMCs or CD71− cell line) for 2 hours at 4°C. After washing to eliminate unbound phages, cells were stained with CD38-fluorescein isothiocyanate (FITC) and CD3-phycoerythrin (PE) or CD3-FITC and CD38-allophycocyanin (all antibodies from Beckmann-Coulter, Hialeah, FL). Target cells and control cells were sorted using FACSAria II (BD Biosciences, Franklin Lakes, NJ) and TG1 E coli were reinfected with phages bound to the sorted cells. Phage output was determined and used for NGS and phage amplification for the next selection round. After the third selection round, single clones were picked, identified with Sanger sequencing (GATC), and scFv phages were produced from selected single clones. To detect binding of scFv phages on myeloma cell lines and control cells, 1 × 1011 phages were incubated with 1 × 106 cells and cell-bound phages were detected with mouse anti-M13 horseradish peroxidase (GE Healthcare) and anti-mouse IgG FITC (Santa Cruz Biotechnology, Dallas, TX) and analyzed by flow cytometry.
NGS guidance of antibody library selections
Variable regions of heavy chains were amplified by PCR using the primers listed in supplemental Table 1 (available on the Blood Web site) at each selection round. Sample-specific barcodes were attached in a second PCR step. PCRs were carried out with the Phusion High-Fidelity DNA Polymerase (NEB). NGS was performed on a MiSeq desktop sequencer (Illumina, San Diego, CA) and data were analyzed with IMGT/HighV-Quest statistics. Complementarity determining region 3 (CDR3) sequences of the most abundant antibody clones bound on myeloma cell lines were plotted over the selection rounds using R statistical software tools (version 3.1.1, license GPL-2).
Determination of HSP translocation by flow cytometry
The Amo-1 myeloma cell line and healthy PBMCs were treated with 10 nM bortezomib (Velcade; Millennium Pharmaceuticals, Cambridge, United Kingdom) for 24 hours and heat-shock protein (HSP) translocation was analyzed by flow cytometry using the following antibodies: mouse anti-HSP60 (Thermo Fisher Scientific, Wilmington, DE), anti-mouse IgG FITC (Santa Cruz Biotechnology), rat anti-HSP71 PE (Abcam, Cambrigde, United Kingdom), rat IgG2a PE isotype (eBioscience, San Diego, CA), rabbit anti-HSP90 (Abcam), and anti-rabbit IgG FITC (Abcam). Data were analyzed using CellQuest Pro software (version 5.2.1; BD Biosciences, Franklin Lakes, NJ).
Results
Exploring the landscape of immunogenic epitopes in myeloma
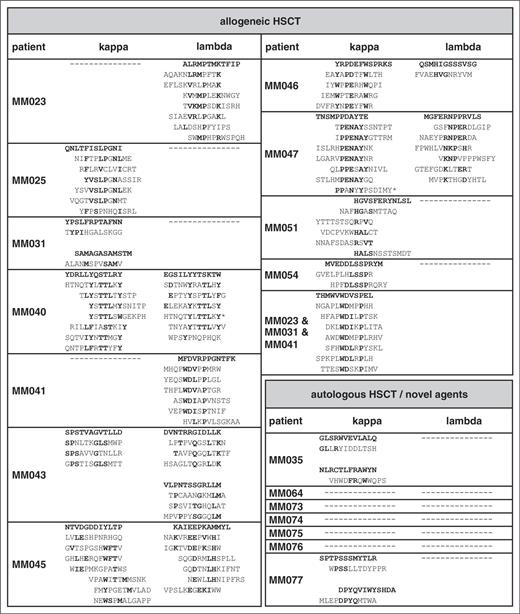
A total of 18 myeloma patients in remission with evidence of oligoclonal antibody bands on immunofixation after treatment were included in this study. Of these, 11 patients had undergone reduced-intensity conditioning allogeneic HSCT and 7 patients had received novel agents (bortezomib or lenalidomide) followed in all but 1 patient by melphalan high-dose chemotherapy with autologous stem cell support (Table 1). For epitope profiling, serum IgG antibodies were purified, separated into κ and λ fractions to reduce the complexity of the oligoclonal antibody spectrum, and screened with a commercial 12-mer random peptide phage display library as schematically shown in supplemental Figure 1A. Two screening approaches were used: first, all 36 IgG κ and λ fractions were screened separately over 3 rounds to obtain epitope-mimics recognized by the most dominant antibody clones of each individual patient. Second, we performed sequential screenings on the antibody repertoires of 3 different patients (MM023, MM031, and MM041) that had been found to be particularly reactive with myeloma antigens in a previous study35 in an attempt to identify epitopes most commonly targeted in different patients. Screenings were successful in all 11 patients after allogeneic HSCT, showing gradual enrichment of antibody-reactive phages over the selection rounds (data not shown). Epitope-mimicking single-phage clones from the third selection rounds of both screening approaches bound the antibody fraction(s) used for selection (exemplarily shown in Figure 1A) and this binding could be blocked by the respective recombinant epitope-mimic in a dose-dependent manner, arguing for the specificity of this interaction (Figure 1B). Insert sequences of all specifically binding phages are displayed in Table 2. Interestingly, between 1 and 3 different unambiguous consensus motifs were found per selection, whereas screenings of healthy donor-derived antibody repertoires yielded specifically binding phage clones lacking an intelligible consensus motif (data not shown). This suggested that up to 3 dominant antibody clones were present in the respective patient antibody repertoire whereas the polyclonal nature of the healthy repertoire prevented enrichment of defined motifs. Also, the sequential screening on the antibody repertoires of patients MM023, MM031, and MM041 yielded a strong consensus motif identical to the motif enriched on MM041 alone, implying that antibodies with this specificity are common in myeloma patients after allogeneic HSCT and particularly abundant in patient MM041. Compared with the allogeneic setting, selections in the autologous setting yielded consensus motifs in a smaller percentage of patients (2 of 7), despite comparably oligoclonal antibody repertoires (Table 2).

Probing serum transplant immunomes by epitope profiling. (A) Binding of individual epitope-mimicking phage clones to serum antibodies of myeloma patients MM023, MM031, and MM041 in clinical remission as well as to polyclonal control IgG. A nonselected random peptide phage was used as a control. Phage binding was measured by ELISA. (B) Inhibition of phage-antibody interaction by cognate peptide. GST-THMWVWDVSPEL blocks the binding of phage THMWVWDVSPEL to MM031 antibodies. Naked GST was used as a control. (C) Epitope-directed serum reactivity in the pre- and post-HSCT period. Here, we show reactivity against epitope-mimic THMWVWDVSPEL with serum antibodies of patient MM031 and of epitope-mimic ALRMPTMKTFIP with serum antibodies of patient MM023 at different time points before and after allogeneic HSCT. (D) Cross-reactivity of epitope-mimics (selected on serum transplant immunomes) with sera from myeloma patients (before and after allogeneic HSCT), healthy donors, and other hematological patients after allogeneic HSCT. A color key was used to depict binding strength in the heat map. Crosses (×) indicate the selections from which each mimic derives. Ab, antibody; CP, control patient (patients who underwent HSCT due to other hematological diseases); HD, healthy donor; MM, MM patient.
Probing serum transplant immunomes by epitope profiling. (A) Binding of individual epitope-mimicking phage clones to serum antibodies of myeloma patients MM023, MM031, and MM041 in clinical remission as well as to polyclonal control IgG. A nonselected random peptide phage was used as a control. Phage binding was measured by ELISA. (B) Inhibition of phage-antibody interaction by cognate peptide. GST-THMWVWDVSPEL blocks the binding of phage THMWVWDVSPEL to MM031 antibodies. Naked GST was used as a control. (C) Epitope-directed serum reactivity in the pre- and post-HSCT period. Here, we show reactivity against epitope-mimic THMWVWDVSPEL with serum antibodies of patient MM031 and of epitope-mimic ALRMPTMKTFIP with serum antibodies of patient MM023 at different time points before and after allogeneic HSCT. (D) Cross-reactivity of epitope-mimics (selected on serum transplant immunomes) with sera from myeloma patients (before and after allogeneic HSCT), healthy donors, and other hematological patients after allogeneic HSCT. A color key was used to depict binding strength in the heat map. Crosses (×) indicate the selections from which each mimic derives. Ab, antibody; CP, control patient (patients who underwent HSCT due to other hematological diseases); HD, healthy donor; MM, MM patient.
Next, we wished to determine the dynamics of epitope recognition over time in patients with longitudinal follow-up. Such time-course experiments suggested that epitopes are only transiently targeted by patient-derived antibodies with a peak within the first year after therapy (Figure 1C).
Disease specificity and clinical relevance of epitope profile
Our data suggested that dominant antibody clones targeting a specific landscape of epitopes were transiently present in the sera of some myeloma patients after therapeutic interventions. This prompted us to specifically address whether: (1) these epitopes were truly disease-specific tumor neoepitopes, (2) overlapping epitope spectra were targeted in different patients, and (3) high epitope reactivity correlated with a favorable response to treatment. We therefore explored the cross-reactivity of epitope-mimics with antibody repertoires from our cohort of myeloma patients after allogeneic HSCT and compared it to the reactivity with antibody repertoires of myeloma patients at first diagnosis, after bortezomib induction and with those of patients with unrelated diseases after allogeneic HSCT and healthy donor repertoires. From each consensus motif, the epitope-mimic showing highest binding to the parental antibody repertoire was chosen as representative clone (bold sequences shown in Table 2). Although all selected epitope-mimics showed highest reactivity with the antibody repertoires they were selected on, they displayed also a considerable degree of cross-reactivity with other antibody repertoires of the myeloma cohort, indicating that recurrent epitopes are targeted in different patients (Figure 1D; supplemental Figure 2). As expected, the epitope-mimic THMWVWDVSPEL identified by the sequential screening approach on different patient antibody repertoires showed the broadest reactivity profile within the cohort. Remarkably, none of the epitope-mimics were cross-reactive with the control antibody repertoires (Figure 1D). Particularly, the lack of cross-reactivity with antibody repertoires from nonmyeloma patients after allogeneic HSCT suggested to us that these epitope-mimics are truly myeloma-specific and do not correspond to epitopes of ubiquitous auto- or infectious antigens. Patients at first diagnosis and after induction treatment did not show reactivity of their sera with our set of epitope-mimics, suggesting that the immunological context of allogeneic HSCT markedly facilitates these B-cell responses. When epitope recognition profiles were correlated with response to therapy 1 year after HSCT, we noted that cases with highly reactive epitope profiles (reactivity with 3 or more epitope-mimics) showed a significantly superior response to treatment (complete vs incomplete remission) compared with cases with low epitope reactivity (Table 1; χ2, P = .007). As expected, the epitope-mimics selected on serum antibodies from patients that had not undergone allogeneic HSCT were less disease-specific and showed a higher degree of cross-reactivity with nonmyeloma immune repertoires (supplemental Figure 3). Together, our epitope profiling suggests common principles in tumor antigen recognition between individual myeloma patients and defines a landscape of immunogenic myeloma-specific epitopes.
Using highly epitope-reactive transplant immunomes as a platform for identification of myeloma cell surface–binding antibodies
Next, we wished to mine highly epitope-reactive patient-derived immunomes for individual antibody sequences with exclusive myeloma cell surface reactivity. Therefore, peripheral blood– or bone marrow–derived B-lineage V-gene immune repertoires from 4 patients in remission after allogeneic HSCT (transplant immunome libraries 1-4) and 1 healthy donor (healthy donor library) were PCR-amplified and cloned as scFv phage display libraries (Figure 2A). These transplant immunome and healthy donor scFv libraries were then positively selected on myeloma cell lines and negatively selected on healthy donor PBMCs or cell lines derived from other malignancies using fluorescence-activated cell sorting as schematically shown in supplemental Figure 1B. As proof-of-principle, a cell surface–binding control phage clone directed against CD71 was spiked into the healthy donor library and selected on a CD71-positive cell line. This resulted in enrichment of binding phage clones over the selection rounds (Figure 2B). After 3 selection rounds, 9 of 10 single clones were identified as the anti-CD71 spike-in clone by Sanger sequencing confirming validity of the experimental setup. For the selections of the transplant immunome libraries, we used a NGS approach to deeply monitor clonal dynamics over the selection rounds. Most of the myeloma transplant immunome selections did not result in enrichment of myeloma-specific clones, suggesting that many of the targeted antigens may in fact not be accessible to the immune system in untreated myeloma cells (exemplarily shown in Figure 2C left panel). Some of the transplant immunome selections, however, led to a continuous enrichment of myeloma cell surface–interacting clones over the selection rounds (exemplarily shown in Figure 2C right panel). This exemplary selection led to a scFv phage clone with heavy-chain CDR3 sequence CARRSDAFDIW, which bound to myeloma cell line EJM, but not to Jurkat control cells (Figure 2D). Interestingly, the most significant enrichments were found for transplant immunome library 4, the only library derived from myeloma patient bone marrow in clinical remission.

Selection of transplant immunome displaying scFv phage libraries on myeloma cells. (A) Schematic diagram illustrating the strategy to clone patients’ B-cell immune repertoires into the pHAL14 scFv phage display vector. (B) Proof-of-principle selection of healthy donor scFv phage library spiked with 0.1% anti-CD71 scFv phage on CD71+ cells. (C) Deep-sequencing–assisted screening of patient-derived transplant immunome scFv phage libraries on myeloma cell lines. Graphs display heavy-chain CDR3 clonal dynamics of the most frequent clones during selection rounds. Smaller clones are summarized in gray. (D) Flow cytometry–binding analysis of scFv phage clone CARRSDAFDIW derived from transplant immunome library 4 on myeloma cell line (EJM) and control cell line (Jurkat). VH, heavy chain variable region; VL, light chain variable region.
Selection of transplant immunome displaying scFv phage libraries on myeloma cells. (A) Schematic diagram illustrating the strategy to clone patients’ B-cell immune repertoires into the pHAL14 scFv phage display vector. (B) Proof-of-principle selection of healthy donor scFv phage library spiked with 0.1% anti-CD71 scFv phage on CD71+ cells. (C) Deep-sequencing–assisted screening of patient-derived transplant immunome scFv phage libraries on myeloma cell lines. Graphs display heavy-chain CDR3 clonal dynamics of the most frequent clones during selection rounds. Smaller clones are summarized in gray. (D) Flow cytometry–binding analysis of scFv phage clone CARRSDAFDIW derived from transplant immunome library 4 on myeloma cell line (EJM) and control cell line (Jurkat). VH, heavy chain variable region; VL, light chain variable region.
HSPs translocated as a result of immunogenic cell death are common targets of the immunome
The apparent inaccessibility of many epitopes prompted us to determine the parental antigens mimicked by the myeloma-specific epitope fingerprint. Unfortunately, none of the epitope-mimics could be reliably traced back to its parental antigen by simple in silico alignment with protein databases, suggesting conformational mimicry. We therefore used a reverse immunization approach as proof-of-principle to identify the parental antigen mimicked by 1 important representative peptide. The epitope-mimic THMWVWDVSPEL was chosen because it was recognized by antibody repertoires of >50% of myeloma patients after allogeneic HSCT. To generate an anti-THMWVWDVSPEL serum, 2 rats were immunized subcutaneously with the phage clone displaying the peptide THMWVWDVSPEL (schematically shown in Figure 3A). The phage context served as a natural adjuvant for immunization. Sera of immunized rats subsequently showed reactivity with the recombinantly expressed peptide, indicating the induction of epitope-specific rat antibodies (Figure 3B-C). The immunized serum was then used to immunodetect the parental myeloma antigen from myeloma protein extracts subjected to 2-dimensional gel electrophoresis (Figure 3D). Only 1 spot was differentially detected by the postimmunization serum. This spot was identified as HSP60 by mass spectrometry.

Reverse immunization approach to trace back epitope-mimic to parental antigen. (A) Schematic diagram of the strategy used to immunize rats for the generation of THMWVWDVSPEL epitope-directed antisera used for target identification. (B-C) Reactivity of rat anti-THMWVWDVSPEL serum with GST-THMWVWDVSPEL by ELISA and western blot. (D) Antigen identification via SERPA technology. Antigen was identified from IM-9 myeloma cell lysate after 2D gel electrophoresis and blotting using rat anti-THMWVWDVSPEL serum as detection antibody. The detected spot was excised and identified by mass spectrometry.
Reverse immunization approach to trace back epitope-mimic to parental antigen. (A) Schematic diagram of the strategy used to immunize rats for the generation of THMWVWDVSPEL epitope-directed antisera used for target identification. (B-C) Reactivity of rat anti-THMWVWDVSPEL serum with GST-THMWVWDVSPEL by ELISA and western blot. (D) Antigen identification via SERPA technology. Antigen was identified from IM-9 myeloma cell lysate after 2D gel electrophoresis and blotting using rat anti-THMWVWDVSPEL serum as detection antibody. The detected spot was excised and identified by mass spectrometry.
Interestingly, identification of HSP60 as parental antigen of the highly reactive epitope-mimic THMWVWDVSPEL fitted well with our previous proteomic study that revealed HSPs from the HSP90 and HSP70 families as well as HSP60 as common antigens targeted by myeloma patient-derived B-cell responses.35 We therefore wished to find out under which circumstances these primarily cytoplasmic proteins were made accessible to the immune system. Based on previously published data on HSP90 cell surface translocation, we hypothesized that these HSPs could be exposed after treatment with drugs causing immunogenic cell death.40 To experimentally address this, we performed bortezomib treatments with myeloma cell lines and measured HSP60, HSP71, and HSP90 translocation to the cell surface by flow cytometry (Figure 4). Interestingly, bortezomib triggered considerable HSP translocation to the cell surface, suggesting that these common targets of the antimyeloma B-cell immunome may be primarily exposed in the context of immunogenic cell death.

HSPs as targets of the transplant immunome are translocated to the myeloma cell surface translocation after bortezomib treatment. Amo-1 myeloma cells were treated with 10 nM bortezomib for 24 hours followed by flow cytometric staining with HSP60-, HSP71-, and HSP90-directed antibodies (B). Untreated cells served as control (A). Isotype antibody or secondary antibody only served as staining control.
HSPs as targets of the transplant immunome are translocated to the myeloma cell surface translocation after bortezomib treatment. Amo-1 myeloma cells were treated with 10 nM bortezomib for 24 hours followed by flow cytometric staining with HSP60-, HSP71-, and HSP90-directed antibodies (B). Untreated cells served as control (A). Isotype antibody or secondary antibody only served as staining control.
Discussion
Apart from the results of a few SEREX (serological identification of antigens by recombinant expression cloning)–based studies,41,42 B-cell antitumor immunity is insufficiently understood in myeloma compared with the considerable amount of data on T-cell–mediated immunity in this disease.25,43-46
Here, we profiled epitope recognition in a cohort of 18 patients after allogeneic or autologous HSCT or novel agent therapy, revealing strikingly disease-specific and overlapping patterns of epitope reactivity. All of these patients showed oligoclonal serum antibodies on immunofixation, suggesting ongoing B-cell immune responses. Yet, the cohort was heterogeneous concerning their cellular immune context with or without immunosuppressive treatment and different intervals between HSCT and blood sampling. We used a synthetic random peptide phage display approach covering >109 different displayed sequences to globally screen and compare epitope reactivities between patients. The phage screenings involve multiple rounds of affinity selection of the phage library on immobilized patient-derived antibodies followed by identification of epitope-mimicking sequences. This technology allows mimicking of almost all antigenic-binding partners, linear, conformational, or even posttranslational modifications, by the phage displayed 12-mer peptides. It is therefore more suitable for global immune repertoire comparisons between patients compared with a SEREX-based approach, where only a fraction of all potential binding partners may be properly presented in the phage context. Our compelling data suggest that almost all patients after allogeneic HSCT, but only a minority of patients after autologous HSCT or after pharmacotherapy with novel compounds, develop such B-cell responses. This clear difference between autologous and allogeneic HSCT may have resulted from functional differences in patient- vs donor-derived B cells or generally a more permissive immune environment in the allogeneic setting.47 Moreover, high serum epitope reactivity correlated significantly with a patient’s ability to achieve a complete remission, indicating that such B-cell immune responses may be prognostically favorable. Loss of antibody titer over time in patients with continued complete remission is likely due to the lowering myeloma burden and therefore antigenic load, resulting in a lesser trigger for antibody production. One common epitope-mimic that was recognized by serum antibodies of >50% of myeloma patients after allogeneic HSCT was then traced back to its parental antigen HSP60. This, together with our previous data from a proteomic study, clearly suggested that HSPs might represent common targets of the myeloma transplant immunome in such patients.35 This (certainly not myeloma-specific) family of proteins may be translocated to the myeloma cell surface in the context of immunogenic cell death.40,48 This might make these proteins accessible to the immune system, although our own preliminary unpublished data failed to demonstrate a role for aberrant posttranslational modification potentially promoting their immunogenicity (data not shown).
The active cell surface exposition of these proteins led us to speculate about the pathophysiological implications of our findings. Immunogenic cell death is a drug-induced process,49-52 first described after doxorubicin treatment in 2005 by Casares et al.53 The drug most known for its ability to induce immunogenic cell death in the context of myeloma treatment is bortezomib40,48,54,55 (used in 17 of 18 patients in our cohort), but other drugs used in our myeloma-conditioning regimens prior to allogeneic HSCT, such as melphalan56-58 and cyclophosphamide,59,60 can act in this way. After treatment, the dying tumor cells secrete or translocate “danger-associated molecular patterns” (DAMPs) to the cell surface. Thereby they recruit antigen-presenting cells such as dendritic cells (DCs), and stimulate the uptake, processing, and presentation of these antigens.61-63 The DCs in turn foster interleukin-1β– and interleukin-17–dependent T-cell activation, which leads to interferon γ (IFNγ)-mediated antimyeloma immune responses.62 If abundant HSP-targeting antibodies opsonize tumor cells in the process of immunogenic cell death, this could in turn facilitate uptake by and activation of DCs, which might then enhance the immunostimulatory effects on T cells potentially leading to prolonged clinical remissions (schematically depicted in Figure 5). The favorable prognosis associated with such B-cell immune responses34 may therefore not primarily rely on antibody effector function itself, but may be mediated indirectly by enhancing myeloma-directed T-cell immunity.64 This notion is supported by recent studies suggesting that myeloma patients may develop HSP-directed T-cell immune responses.44,65

Schematic representation of potential T-cell activation by anti-HSP–directed humoral immune responses. In the context of immunogenic cell death, HSPs are secreted or translocated to the cell surface and serve as DAMPs. Such DAMPs can recruit and activate DCs directly as shown on the right. Opsonization of HSPs by the antibody immune response may even enhance this DC activation by interaction of immune complexes with the Fcγ receptor (left part). Both ways may result in engulfment of DAMPs and potent tumor antigen presentation to T cells, potentially leading to effective T-cell–mediated antimyeloma immune responses. CTL, cytotoxic T-lymphocytes. Diagram adapted from Kroemer et al.62
Schematic representation of potential T-cell activation by anti-HSP–directed humoral immune responses. In the context of immunogenic cell death, HSPs are secreted or translocated to the cell surface and serve as DAMPs. Such DAMPs can recruit and activate DCs directly as shown on the right. Opsonization of HSPs by the antibody immune response may even enhance this DC activation by interaction of immune complexes with the Fcγ receptor (left part). Both ways may result in engulfment of DAMPs and potent tumor antigen presentation to T cells, potentially leading to effective T-cell–mediated antimyeloma immune responses. CTL, cytotoxic T-lymphocytes. Diagram adapted from Kroemer et al.62
Despite the fact that all of our patients have been treated with inducers of immunogenic cell death prior to serum antibody analysis, only those who underwent allogeneic HSCT (but not those at first diagnosis, after induction or after autologous HSCT) show a high frequency of anti-HSP or antimyeloma B-cell immune responses. One potential explanation for the more B-cell permissive immune environments after allogeneic HSCT could be related to T-cell exhaustion in the context of myeloma and autologous HSCT because B cells need T-cell help in the majority of responses. T-cell exhaustion is prognostically unfavorable,66-68 may not be reversed by autologous HSCT,67 and clinical studies targeting this exhaustion by immune checkpoint inhibition after autologous HSCT are under way.69,70 Also, our own preliminary data point in this direction, since we found exhausted programmed cell death protein 1 (PD-1) expressing CD3+ circulating T cells to be clearly expanded after autologous HSCT as compared with the allogeneic setting (data not shown). This finding may represent a potential explanation as to why the allogeneic setting produces more specific and more frequent antimyeloma B-cell responses.
The common therapy-modulated HSP antigenic targets add to our understanding of B- and T-cell immunology in the autologous and allogeneic setting, but may not be good therapeutic targets due to unstable expression. However, we also provide proof-of-principle that myeloma-interacting (lower frequency) antibody clones targeting stably expressed surface structures may be identified on our platform under stringent selection conditions. This novel type of NGS-assisted discovery platform introduced here may therefore be used in the development of novel immunotherapeutic approaches in myeloma and potentially other cancers with disease-specific B-cell immune responses.71 It represents an attractive, state-of-the-art technology for antibody screening due to its short turnaround time, low costs, and low experimental effort because the NGS technology obviates the need for evaluation of large numbers of single clones.72 Moreover, antibodies arising from this platform are of human origin and therefore do not have to be humanized for clinical applications.73 Our data also show that even if only a small fraction of antibodies from a patient’s immune repertoire react strongly with the cancer cell surface, stringent selection conditions may allow for amplification of such rare clones, provided that the diversity of the library covers many, if not all, conceivable combinations of heavy and light chains present in the fraction of the immune repertoire that was used for library construction. Our results also suggest that certain immune environments are more suitable than others, with the bone marrow–derived transplant immunome showing better results than immunomes derived from peripheral blood of transplanted myeloma patients.
Taken together, our B-lineage immunoprofiling reveals a myeloma-specific epitope fingerprint, which opens up new pathophysiological perspectives in patients undergoing autologous and allogeneic HSCT. Moreover, the NGS-driven technology presented here may be exploited in the development of novel immunotherapeutic options in myeloma and potentially other diseases. This comprises monoclonal antibodies, bispecific T-cell engagers, chimeric antigen receptor T-cell technology (relying on scFv-mediated antigen recognition), and neoepitope vaccination strategies, which might become increasingly important in light of the recent advances in immune checkpoint inhibition.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Anita Schulenkorf, Arne Düsedau, Fabienne Seyfried, and Gudrun Dubberke (University Medical Center Hamburg-Eppendorf Antibody Core Unit) for excellent technical support and Dominik Thiele for the heat-map design.
This work was supported by grant 110906 (M.B.) from the German Cancer Aid. M.B. holds a professorship for immunological cancer research and therapy supported by the Hubertus Wald-Foundation, Hamburg.
Authorship
Contribution: M.B. designed the study, supervised the experiments, and wrote the manuscript; A.S. performed the HSP translocation experiments, analyzed clinical data, and contributed to the writing of the manuscript; A.O. performed the scFv screenings and NGS preparation and contributed to the writing of the manuscript; B.T. established the scFv cloning and phage display, computed the bioinformatics, and plotted the NGS data; F. Hofmann, M.G., and M.V. performed random peptide phage selections; M.H. and S.M. provided the scFv library cloning vectors and strategy and participated in library cloning; F.B., F. Haag, and F.K.-N. prepared and conducted the rat immunization and target identification; D.I. and A.G. performed the NGS; M.A. performed the bioinformatics; C.B. interpreted data and critically revised the manuscript; U.B., U.-M.v.P., and N.K. provided patient material and clinical data; and all authors reviewed the manuscript and approved the final version.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Mascha Binder, Department of Oncology and Hematology, University Medical Center Hamburg-Eppendorf, Martinistrasse 52, D-20246 Hamburg, Germany; e-mail: m.binder@uke.de.
