

Key Points
Association of platelet Clec-2 with lymphatic podoplanin regulates lung development by facilitating alveolar duct myofibroblast differentiation.
Alveolar duct myofibroblast differentiation is regulated in part by transforming growth factor-β released from platelets activated by Clec-2/podoplanin interaction.

Abstract
Platelets participate in not only thrombosis and hemostasis but also other pathophysiological processes, including tumor metastasis and inflammation. However, the putative role of platelets in the development of solid organs has not yet been described. Here, we report that platelets regulate lung development through the interaction between the platelet-activation receptor, C-type lectin-like receptor-2 (Clec-2; encoded by Clec1b), and its ligand, podoplanin, a membrane protein. Clec-2 deletion in mouse platelets led to lung malformation, which caused respiratory failure and neonatal lethality. In these embryos, α-smooth muscle actin-positive alveolar duct myofibroblasts (adMYFs) were almost absent in the primary alveolar septa, which resulted in loss of alveolar elastic fibers and lung malformation. Our data suggest that the lack of adMYFs is caused by abnormal differentiation of lung mesothelial cells (luMCs), the major progenitor of adMYFs. In the developing lung, podoplanin expression is detected in alveolar epithelial cells (AECs), luMCs, and lymphatic endothelial cells (LECs). LEC-specific podoplanin knockout mice showed neonatal lethality and Clec1b−/−-like lung developmental abnormalities. Notably, these Clec1b−/−-like lung abnormalities were also observed after thrombocytopenia or transforming growth factor-β depletion in fetuses. We propose that the interaction between Clec-2 on platelets and podoplanin on LECs stimulates adMYF differentiation of luMCs through transforming growth factor-β signaling, thus regulating normal lung development.
Introduction
In mammals, air inflation into the lungs immediately after birth is one of the most important events for survival. Lung development is a hierarchical process involving branching morphogenesis, alveolar septum formation, and surfactant secretion; the precise execution of this process is indispensable for lung respiration. Lung development is divided into 5 phases: embryonic, pseudoglandular, canalicular, saccular, and alveolar. During the canalicular and saccular phases (E16.5-P0 in mice [E, embryonic day; P, postnatal day]), drastic morphological changes are observed in the distal lung, which collectively lead to alveolar sac formation, also termed primary septum formation.1-3
α-smooth muscle actin-positive alveolar duct myofibroblasts (adMYFs) are detected in the interstitium between alveolar sacs.3,4 adMYFs secrete extracellular matrix (ECM) proteins, such as elastin and collagen, which contribute to the normal dilation of alveolar sacs and afford mechanical strength, flexibility, and elasticity to alveoli.4-7 Developmentally, it is thought that lung mesothelial cells (luMCs) contribute to the adMYF population8 and that multiple signaling pathways are involved in adMYF differentiation.4,5,9
There is increasing evidence that platelets have important roles not only in thrombosis and hemostasis but also in various pathophysiological situations. C-type lectin-like receptor-2 (Clec-2) is a multifaceted platelet activation receptor that functions in blood/lymphatic-vessel separation, vasculature integrity maintenance, megakaryopoiesis, and thrombus formation.10,11 In mice, Clec-2 is highly expressed in platelets and megakaryocytes, but it is also weakly expressed in certain white blood cells.12,13 The endogenous Clec-2 ligand is podoplanin (Pdpn),14 which is a membrane protein expressed on multiple cell types, such as lymphatic endothelial cells (LECs), type I alveolar epithelial cells (AEC1), and kidney podocytes.15 Recently, platelets have been considered to be a source of various bioactive substances, such as growth factors, small molecules, and lipid mediators.16-19 For example, sphingosine-1-phosphate released from platelets by Clec-2/Pdpn-dependent platelet activation maintains the vascular integrity of high endothelial venules in lymph nodes.20
In general, proper organogenesis proceeds through highly regulated interactions among different tissues within and/or of neighboring organs. To our knowledge, no study has reported the involvement of circulating platelets in the normal development of solid organs. In the present study, we performed detailed phenotypic analyses of the fetal lung of mice lacking Clec-2/Pdpn signaling molecules to identify the putative role of platelets in lung development. Our findings propose a novel mechanism of the critical role of platelets in lung development: the interaction between Clec-2 on platelets and Pdpn on LECs stimulates luMC differentiation into adMYFs through transforming growth factor (TGF)-β signaling, thus regulating normal lung development.
Materials and methods
Mice
All animal procedures in this study were approved by the animal care and use committee of the University of Yamanashi, and all animal experiments were conducted in compliance with ethical guidelines and the approved protocols. Mouse strains used in this study are described in the supplemental Methods, available on the Blood Web site. The appropriate strains were used to set up matings performed to collect embryos at the appropriate stages, depending on their use.
Evaluation of neonatal lethality and genotyping
Naturally born neonates at P0 were collected, the live and dead neonates were counted, and genotyping was performed by using the primers listed in supplemental Table 1.
Tissue preparation and histological analyses
Fetal lungs were fixed in 4% paraformaldehyde in phosphate-buffered saline. For hematoxylin and eosin or Elastica van Gieson staining, samples were embedded in paraffin and sectioned at a thickness of 5 μm. For immunohistochemistry, samples were embedded in O.C.T. compound (Sakura Finetek, Japan), and sectioned at a thickness of 10 μm. Whole-mount immunohistochemistry was performed according to the procedure available on the Abcam website. Supplemental Table 2 lists the primary and secondary antibodies used for immunostaining. Samples were imaged using a fluorescence microscope (IX-71; Olympus, Japan) or a laser confocal microscope (LSM-10; Olympus).
Analysis of Clec-2 expression on fetal platelets
Fetal blood at E17.5 was collected in 5 mM EDTA in phosphate-buffered saline immediately after decapitation, and platelet-rich supernatants were obtained after centrifugation. Platelets were stained using antibodies listed in Supplemental Table 2. Co-staining with anti-CD41–Alexa Fluor 546 was performed to gate on platelets. Clec-2 expression was measured by using a BD Accuri C6 flow cytometry system (BD Biosciences). The value obtained after subtracting the isotype mean fluorescence intensity from the Clec-2 mean fluorescence intensity was defined as the Clec-2 expression level.
Antibody treatment of fetuses
Anti-Clec-2 was intraperitoneally injected into pregnant mice at E10.5 and E14.5 (10 μg/g for each injection). Anti-GPIb was injected at E10.5 and E14.5 (5 μg/g for each injection), and anti-TGF-β was injected at E14.5 (50 μg/body). Rat or mouse immunoglobulin G (IgG) was used as a control. Antibody-treated embryos were collected at the appropriate stages, depending on their use in the after experiments.
Quantification of TGF-β1
To prepare lung lysates, fetal lungs at E17.5 were minced in radioimmunoprecipitation assay buffer and lysed by sonication. The concentrations of TGF-β1 in appropriately diluted lysates were measured using enzyme-linked immunosorbent assay kits for TGF-β1 (Thermo Fisher Scientific), following the manufacturers’ instructions.
Statistical analysis
Statistical analyses were appropriately performed using Fisher’s exact test in js-STAR (version 8.0.1j, Satoshi Tanaka and nappa [Hiroyuki Nakano]) or unpaired Student t test, Holm-Sidak test, Tukey’s multiple-comparison test, or Dunnett’s multiple-comparison test in GraphPad Prism 6.
Results
Clec1b-deficient mice show complete neonatal lethality, abnormal lung morphology, and elastogenesis
Systemic Clec1b-deficient (Clec1b−/−) mice exhibit complete neonatal lethality21,22 (Figure 1A); however, the cause of death has not been established. Clec1b+/+ neonates started breathing activity immediately after caesarean section and became pink by sufficient oxygenation within 5 to 10 minutes. Clec1b−/− neonates also started breathing activity (data not shown), but they had a cyanotic appearance and died within 1 hour (Figure 1B). Numerous air bubbles were observed in Clec1b+/+ but not Clec1b−/− neonatal lungs (Figure 1B). The surfaces of Clec1b−/− lung lobes at E17.5 appeared lumpy, although no morphological abnormality was apparent at E16.5 (Figure 1C). At E18.5, marginal regions of Clec1b+/+ lung lobes became thinner and alveolar sacs became visible, whereas these regions remained thick and solid in Clec1b−/− lungs (Figure 1C). Cyanosis, air inflation failure with normal breathing activities, and lung morphological abnormality in Clec1b−/− fetuses suggested that neonatal lethality was a result of lung developmental abnormalities, not defects in neuromuscular function or central respiratory drive.
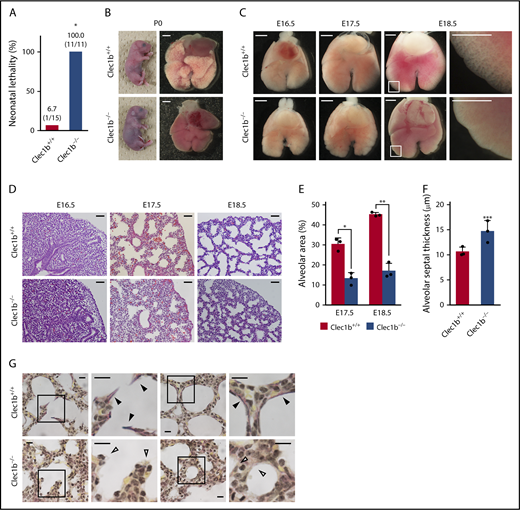
Morphological and histological abnormalities in Clec1b-deficient lung. (A) Neonatal lethality of Clec1b+/+ and Clec1b−/− mice. Numbers within parentheses: dead/total neonates at P0. *P = .0000, Fisher’s exact test. (B) Cyanotic appearance of whole-body and lung air inflation failure in Clec1b−/− neonates. (C) Ventral views of E16.5/E17.5/E18.5 fetal lungs; magnified views of framed areas in images are shown on the right. (D) Hematoxylin and eosin staining of E16.5/E17.5/E18.5 lung sections. (E-F) Quantification of alveolar area (E) and alveolar septum thickness (F); mean ± SD, n = 3 each. *P = .0023; **P = .0003; ***P= .0414, Student t test. (G) Elastica van Gieson staining of P0 lung sections. Magnified views of framed areas in images are shown on the right. Black and open arrowheads: mature elastic fibers (dark purple) and alveolar septa without elastic fibers, respectively. Scale bars: 1 mm (B-C); 25 μm (D); 10 μm (G).
Morphological and histological abnormalities in Clec1b-deficient lung. (A) Neonatal lethality of Clec1b+/+ and Clec1b−/− mice. Numbers within parentheses: dead/total neonates at P0. *P = .0000, Fisher’s exact test. (B) Cyanotic appearance of whole-body and lung air inflation failure in Clec1b−/− neonates. (C) Ventral views of E16.5/E17.5/E18.5 fetal lungs; magnified views of framed areas in images are shown on the right. (D) Hematoxylin and eosin staining of E16.5/E17.5/E18.5 lung sections. (E-F) Quantification of alveolar area (E) and alveolar septum thickness (F); mean ± SD, n = 3 each. *P = .0023; **P = .0003; ***P= .0414, Student t test. (G) Elastica van Gieson staining of P0 lung sections. Magnified views of framed areas in images are shown on the right. Black and open arrowheads: mature elastic fibers (dark purple) and alveolar septa without elastic fibers, respectively. Scale bars: 1 mm (B-C); 25 μm (D); 10 μm (G).
At E17.5, epithelial cells in acinar buds begin to alter their shape from columnar to squamous, and the surrounding interstitium becomes thin to form the primary septa with sufficiently dilated alveolar sacs.1-3 Therefore, we next conducted histological analyses to examine primary septum formation. At E17.5 and E18.5, Clec1b−/− lungs showed a significantly smaller alveolar area (Figure 1D-E) and thicker alveolar septum (Figure 1D,F), although no histological abnormality was observed at E16.5 (Figure 1D). Elastica van Gieson staining revealed mature elastic fibers in Clec1b+/+ alveolar septa, whereas such fibers were rarely detected in Clec1b−/− lung (Figure 1G).
adMYF differentiation is impaired in Clec1b−/− mice
To determine how Clec1b deficiency causes abnormal primary septum formation, we tested the expression of cell type-specific markers at E17.5 by immunohistochemistry. Aqp5 and Pdpn, markers of AEC1; ProSPC, a marker of type II alveolar epithelial cells (AEC2); and CC10, a marker of Clara cells, were expressed normally in the Clec1b−/− lung (Figure 2A). Blood and lymphatic vessel formation in the Clec1b−/− lung appeared normal by CD34 or Lyve-1/Prox1 staining (Figure 2A). In addition, transmission electron microscopy revealed normal lamellar body formation and surfactant secretion in Clec1b−/− lungs (Figure 2B).
![Figure 2. Clec1b deficiency results in absence of adMYFs, but not defective differentiation of other lung cell types, surfactant secretion, and pulmonary edema. (A) Expression pattern of cell-type-specific markers in E17.5 lung. Lung sections of Clec1b+/+ and Clec1b−/− fetuses were stained for markers of AEC1s (aquaporin-5 [Aqp5] and podoplanin [Pdpn]), AEC2s (prosurfactant protein C [ProSPC]), Clara cells (CC10), vascular endothelial cells (CD34), and LECs (Prox1 and Lyve-1) and with DAPI (blue). (B) Transmission electron microscopy images of lamellar bodies in AEC2s (open arrowheads) and secreted surfactants (black arrowheads). (C) Lung wet/dry (W/D) ratio in Clec1b+/+ and Clec1b−/− neonates; mean ± SD, n = 7 and 5, Student t test. N.S., not significant. (D) Immunostaining of α-SMA (green) and DAPI (blue) in E16.5/E17.5 distal lung. Arrowheads: bronchial smooth muscle cells, which are not adMYFs. (E) Quantification of α-SMA expression in E17.5 distal lung. α-SMA staining intensity was measured from the lung surface to a depth of 75 μm, as described in the supplemental Materials; mean ± SD, n = 3 each. *P = .0486, **P = .0053, Holm-Sidak test. AL, alveolar sac; BR, bronchiole; LV, lymphatic vessel; M, lung mesothelium; V, vein. Scale bars: 25 μm (A,D); 2 μm (B).](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/132/11/10.1182_blood-2017-12-823369/4/m_blood823369f2.png?Expires=1765924097&Signature=Kcv4kAgtl5MM67oW7Wto8-clOXS2V-T7CMxVmg91MNb4BHgobcgApsf2rvrwaA8HYdaK2LUGSWpiXX6ZwyX05goLmtKM5ZPu-SIiwLWS5-NoiV0jvvUh9rN9cLgoQiLe7O~PB4ij3LYIsb4zyYpxX4H6RD7sPeZ16cYq5HXQdIiB6No0ElXF26iUAfT-NaAusPmyjt7Vab~5qw1KRFEoWOv3mO2Yg2zgjQLWRm-uzU9nWUneXWtPGf6MuxilMOIrEcVSHM8be~4C6eCY1yU5thznGf6vAiUNQBQro99vNI~JKlI~a39xCfU31Qeu9IuNIxoR2e9irPEtLLu3R5E7~g__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Clec1b deficiency results in absence of adMYFs, but not defective differentiation of other lung cell types, surfactant secretion, and pulmonary edema. (A) Expression pattern of cell-type-specific markers in E17.5 lung. Lung sections of Clec1b+/+ and Clec1b−/− fetuses were stained for markers of AEC1s (aquaporin-5 [Aqp5] and podoplanin [Pdpn]), AEC2s (prosurfactant protein C [ProSPC]), Clara cells (CC10), vascular endothelial cells (CD34), and LECs (Prox1 and Lyve-1) and with DAPI (blue). (B) Transmission electron microscopy images of lamellar bodies in AEC2s (open arrowheads) and secreted surfactants (black arrowheads). (C) Lung wet/dry (W/D) ratio in Clec1b+/+ and Clec1b−/− neonates; mean ± SD, n = 7 and 5, Student t test. N.S., not significant. (D) Immunostaining of α-SMA (green) and DAPI (blue) in E16.5/E17.5 distal lung. Arrowheads: bronchial smooth muscle cells, which are not adMYFs. (E) Quantification of α-SMA expression in E17.5 distal lung. α-SMA staining intensity was measured from the lung surface to a depth of 75 μm, as described in the supplemental Materials; mean ± SD, n = 3 each. *P = .0486, **P = .0053, Holm-Sidak test. AL, alveolar sac; BR, bronchiole; LV, lymphatic vessel; M, lung mesothelium; V, vein. Scale bars: 25 μm (A,D); 2 μm (B).
Clec1b deficiency results in absence of adMYFs, but not defective differentiation of other lung cell types, surfactant secretion, and pulmonary edema. (A) Expression pattern of cell-type-specific markers in E17.5 lung. Lung sections of Clec1b+/+ and Clec1b−/− fetuses were stained for markers of AEC1s (aquaporin-5 [Aqp5] and podoplanin [Pdpn]), AEC2s (prosurfactant protein C [ProSPC]), Clara cells (CC10), vascular endothelial cells (CD34), and LECs (Prox1 and Lyve-1) and with DAPI (blue). (B) Transmission electron microscopy images of lamellar bodies in AEC2s (open arrowheads) and secreted surfactants (black arrowheads). (C) Lung wet/dry (W/D) ratio in Clec1b+/+ and Clec1b−/− neonates; mean ± SD, n = 7 and 5, Student t test. N.S., not significant. (D) Immunostaining of α-SMA (green) and DAPI (blue) in E16.5/E17.5 distal lung. Arrowheads: bronchial smooth muscle cells, which are not adMYFs. (E) Quantification of α-SMA expression in E17.5 distal lung. α-SMA staining intensity was measured from the lung surface to a depth of 75 μm, as described in the supplemental Materials; mean ± SD, n = 3 each. *P = .0486, **P = .0053, Holm-Sidak test. AL, alveolar sac; BR, bronchiole; LV, lymphatic vessel; M, lung mesothelium; V, vein. Scale bars: 25 μm (A,D); 2 μm (B).
A previous report showed that deletion of CCBE1 or Vegfr3 causes lack of lung lymphatics and severe pulmonary edema, which leads to abnormal primary septum formation, low lung compliance, and neonatal lethality.23 Because Clec1b−/− fetuses exhibit subcutaneous edema by blood–lymphatic-vessel misconnection,21,22 we next examined whether Clec1b−/− lungs exhibit pulmonary edema. There was no significant difference in lung wet weight, dry weight, and wet/dry ratio between Clec1b+/+ and Clec1b−/− lungs, whereas Clec1b−/− neonates had significantly higher body weight than Clec1b+/+ neonates, which was probably caused by subcutaneous edema (Figure 2C; supplemental Figure 1A-C). However, we assume that subcutaneous edema did not cause neonatal lethality, as Aspp1−/− mice with severe subcutaneous edema and slight lung edema by impaired lymphatic vessel assembly24 did not show neonatal lethality (supplemental Figure D-E). These findings suggest that neonatal lethality of Clec1b−/− mice is caused by neither pulmonary edema nor subcutaneous edema.
We next assessed the expression of the myofibroblast marker α-SMA. At E16.5, α-SMA expression was hardly detected in the distal lung interstitium in both Clec1b+/+ and Clec1b−/− mice, although bronchial smooth muscle cells, which emerge earlier4,25 than adMYFs, were strongly stained (Figure 2D). At E17.5, α-SMA was detected throughout the distal lung interstitium in Clec1b+/+ fetuses, but only in the submesothelial area and minimally in the interstitium in Clec1b−/− fetuses (Figure 2D), which was confirmed by quantitative analyses, according to distance from the lung surface (Figure 2E). adMYFs are considered to be critical for alveolar septum formation through ECM deposition, because several knockout mouse strains lacking adMYFs (eg, Pdgfa−/−, Lfng−/−, and Gr1−/−) demonstrate alveolar malformation and impaired elastogenesis.4,5,9 Collectively, these results suggested that the lung developmental abnormalities and neonatal lethality in Clec1b−/− mice were caused by lack of adMYFs in the primary septum, and not by defects in epithelial cell differentiation, vascular networks, surfactant secretion, or edema in the lung.
Clec1b deficiency causes hyperproliferation and abnormal differentiation in luMCs
luMCs are considered to differentiate into lung mesenchymal cells, including adMYFs, through mesothelial-to-mesenchymal transition.8,26 To confirm this phenomenon, we conducted adMYF lineage tracing by using Wt1-EGFPCre, CAG-CAT-EGFP mice, in which all luMC-descendant cells constitutively express EGFP.8,26 At E17.5, a majority of the adMYFs were found to be EGFP-positive (Figure 3A), suggesting that luMCs are the major adMYF progenitors and that lack of adMYFs in Clec1b−/− lungs may be caused by some luMC abnormalities. Thus, we next examined luMC proliferation, apoptosis, and differentiation. The fraction of 5-bromo-2′-deoxyuridine (BrdU)-positive proliferating cells was significantly increased in luMCs of Clec1b−/− fetuses (Figure 3B-C). TUNEL-positive apoptotic cells were hardly detected among both Clec1b+/+ and Clec1b−/− luMCs (Figure 3D). To evaluate luMC differentiation, we immunostained luMCs for Wilms’ tumor 1 (Wt1), which is a transcription factor specifically expressed in luMCs during development (Figure 3E).26 Wt1 is known to maintain mesothelial characteristics and prevent myofibroblastic differentiation.26,27 At E15.5 and E16.5, only around 5% luMCs were Wt1-negative in both Clec1b+/+ and Clec1b−/− fetuses (Figure 3F). At E17.5, the rate was significantly increased to approximately 20% in Clec1b+/+, but was not changed in Clec1b−/− fetuses (Figure 3F). The luMC density decreased with time in Clec1b+/+ fetuses, but was maintained at a rather high level in Clec1b−/− fetuses (Figure 3G). Accordingly, we found that luMCs in Clec1b−/− fetuses exhibited hyperproliferative activity (Figure 3B). In E17.5 Wt1-EGFPCre, CAG-CAT-EGFP lungs, all luMCs were labeled with EGFP (Figure 3A), indicating that Wt1 is initially expressed in all luMCs but is subsequently downregulated in a portion of luMCs. These results suggest that appropriate downregulation of Wt1 in a portion of luMCs balances luMC proliferation with differentiation into adMYFs, whereas Clec1b deficiency abnormally maintains Wt1 expression in luMCs, thereby disrupting the balance and causing luMC hyperproliferation and defective differentiation into adMYFs.
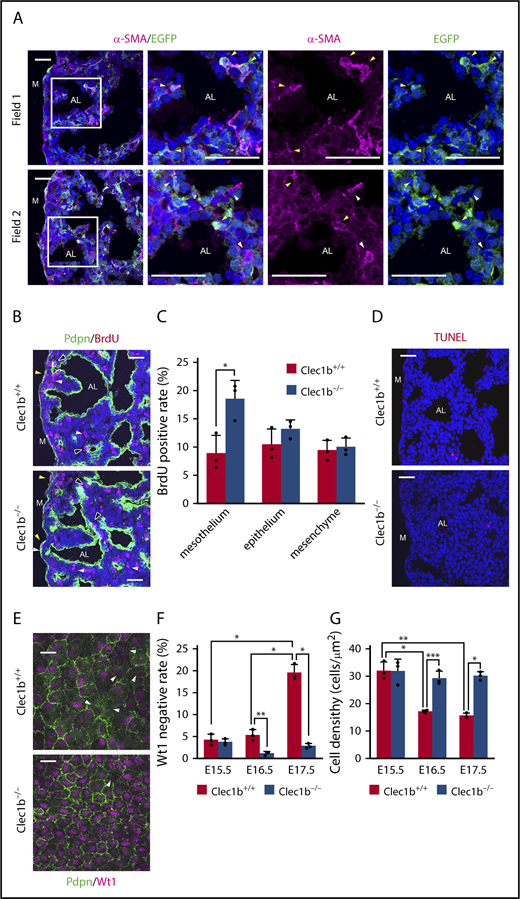
Clec1b deficiency causes lung mesothelial cell hyperproliferation and abnormal differentiation. (A) Lineage tracing of adMYFs in E17.5 Wt1-EGFPCre, CAG-CAT-EGFP lung sections by α-SMA (magenta) and EGFP (green) staining and DAPI (blue) counterstaining; magnified views of framed areas in images are shown on the right. Arrowheads indicate α-SMA+EGFP+ cells (yellow), α-SMA+EGFP− cells (white). (B) Immunostaining of BrdU (red) and Pdpn (green) and DAPI staining (blue) of E17.5 distal lung sections. Pdpn immunostaining distinguishes the mesothelium, epithelium, and mesenchyme, as Pdpn is expressed in luMCs, AECs, and LECs in the distal lung. Arrowheads: BrdU-positive mesothelial cells (yellow), epithelial cells (black), and mesenchymal cells (white). (C) Histogram showing BrdU-positive rates in mesothelial, epithelial, and mesenchymal cells in E17.5 distal lung; mean ± SD, n = 3 each; *P = .0014, Holm-Sidak test. (D) TUNEL (red) and DAPI (blue) staining of E17.5 distal lung sections. TUNEL-positive apoptotic cells were very rare in both Clec1b+/+ and Clec1b−/−. (E) Whole-mount immunohistochemistry of Wt1 (magenta) and Pdpn (green) in E17.5 luMCs. Arrowheads: Wt1-negative luMCs. (F) Quantification of Wt1-negative rate in luMCs at E15.5, E16.5, and E17.5; mean ± SD, n = 3 each. *P < .0001; **P = .0074, Tukey’s test. (G) Quantification of luMC density at E15.5, E16.5, and E17.5; mean ± SD, n = 3 each. *P = .0002; **P < .0001; ***P = .0010, Tukey’s test. AL, alveolar sac; M, lung mesothelium. Scale bars: 25 μm (A-B,D,E).
Clec1b deficiency causes lung mesothelial cell hyperproliferation and abnormal differentiation. (A) Lineage tracing of adMYFs in E17.5 Wt1-EGFPCre, CAG-CAT-EGFP lung sections by α-SMA (magenta) and EGFP (green) staining and DAPI (blue) counterstaining; magnified views of framed areas in images are shown on the right. Arrowheads indicate α-SMA+EGFP+ cells (yellow), α-SMA+EGFP− cells (white). (B) Immunostaining of BrdU (red) and Pdpn (green) and DAPI staining (blue) of E17.5 distal lung sections. Pdpn immunostaining distinguishes the mesothelium, epithelium, and mesenchyme, as Pdpn is expressed in luMCs, AECs, and LECs in the distal lung. Arrowheads: BrdU-positive mesothelial cells (yellow), epithelial cells (black), and mesenchymal cells (white). (C) Histogram showing BrdU-positive rates in mesothelial, epithelial, and mesenchymal cells in E17.5 distal lung; mean ± SD, n = 3 each; *P = .0014, Holm-Sidak test. (D) TUNEL (red) and DAPI (blue) staining of E17.5 distal lung sections. TUNEL-positive apoptotic cells were very rare in both Clec1b+/+ and Clec1b−/−. (E) Whole-mount immunohistochemistry of Wt1 (magenta) and Pdpn (green) in E17.5 luMCs. Arrowheads: Wt1-negative luMCs. (F) Quantification of Wt1-negative rate in luMCs at E15.5, E16.5, and E17.5; mean ± SD, n = 3 each. *P < .0001; **P = .0074, Tukey’s test. (G) Quantification of luMC density at E15.5, E16.5, and E17.5; mean ± SD, n = 3 each. *P = .0002; **P < .0001; ***P = .0010, Tukey’s test. AL, alveolar sac; M, lung mesothelium. Scale bars: 25 μm (A-B,D,E).
Clec-2 on platelets is required for lung development
In adult mice, Clec-2 is expressed on platelets and megakaryocytes, as well as white blood cells such as neutrophils.12,13 We used immunostaining to identify Clec-2-expressing cells during lung development and observed exclusive Clec-2 expression in platelets and megakaryocytes (Figure 4A; supplemental Figure 2). Previously, platelet Clec-2 was not assumed to be involved in lung development and neonatal survival because platelet-specific Clec1b-deficient mice (Pf4-Cre, Clec1bfl/fl, referred to here as Clec1bΔPLT) grew normally to adulthood.28 However, our detailed analyses revealed that Clec1bΔPLT mice exhibit slight lung morphological abnormality (Figure 4B), slightly but significantly higher neonatal lethality (Figure 4C), lower interstitial α-SMA expression (Figure 4D-E), and higher luMC density compared with Clec1bfl/fl (wild-type) mice (Figure 4F-G). Moreover, the Wt1-negative rate was decreased, albeit not significantly, in Clec1bΔPLT mice (Figure 4H). Thus, Clec1bΔPLT mice showed a mild “Clec1b−/−-like” phenotype. Although platelet counts are lower in adult Clec1bΔPLT than in wild-type mice,29 fetal platelet counts did not differ among Clec1b+/+, Clec1b−/−, Clec1bfl/fl, and Clec1bΔPLT mice, which is similar to the reported platelet number in murine wild-type fetuses30 (Figure 4I). Therefore, we suspected that the mild phenotype of Clec1bΔPLT mice resulted from insufficient Clec1b deletion in platelets.
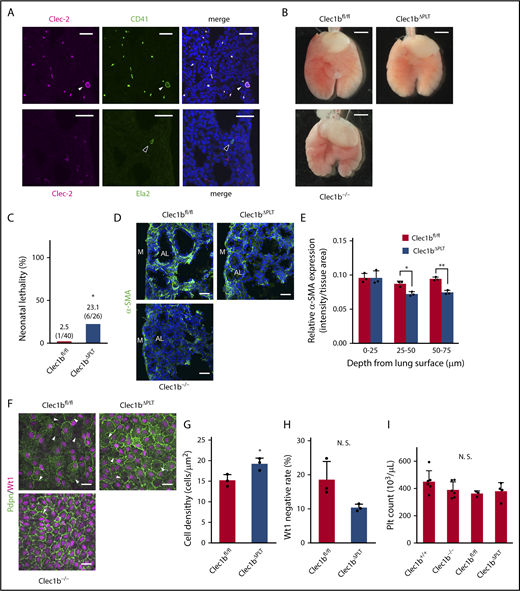
Platelet-specific Clec1b-deficient (Clec1bΔPLT) mice show mild Clec1b−/−-like lung abnormalities. (A) Clec-2 staining (magenta) with CD41 (green, platelets, and megakaryocytes) or Ela2 (green, neutrophils) and DAPI (blue) in E17.5 wild-type lungs. Arrowheads indicate megakaryocytes (white) and neutrophils (black). Clec-2 expression entirely overlapped with CD41 expression, but not with Ela2. (B) Ventral views of Clec1bfl/fl (wild-type), Clec1bΔPLT, and Clec1b−/− fetal lungs at E17.5. (C) Neonatal lethality of Clec1bfl/fl and Clec1bΔPLT neonates. *P = .00127, Fisher’s exact test. (D) Immunostaining of α-SMA (green) and DAPI (blue) in E17.5 distal lung. AL, alveolar sac; M, lung mesothelium. (E) Quantification of α-SMA expression in Clec1bfl/fl and Clec1bΔPLT distal lung at E17.5; mean ± SD, n = 3 each. *P = .0211; **P = .0033, Holm-Sidak test. (F) Whole-mount immunohistochemistry of Wt1 (magenta) and Pdpn (green) in E17.5 luMCs. Arrowheads: Wt1-negative luMCs. (G) Quantification of luMC density; mean ± SD, n = 3 each. *P = .0281, Student t test. (H) Quantification of Wt1-negative rate in luMCs; mean ± SD, n = 3 each. N. S., not significant; P = .062, Student t test. (I) Platelet counts in E17.5 Clec1b+/+, Clec1b−/−, Clec1bfl/fl, and Clec1bΔPLT fetal blood; mean ± SD, n = 6, 6, 3, and 4, respectively. N.S., not significant, Tukey’s test. Scale bars: 25 μm (A,D,F); 1 mm (B).
Platelet-specific Clec1b-deficient (Clec1bΔPLT) mice show mild Clec1b−/−-like lung abnormalities. (A) Clec-2 staining (magenta) with CD41 (green, platelets, and megakaryocytes) or Ela2 (green, neutrophils) and DAPI (blue) in E17.5 wild-type lungs. Arrowheads indicate megakaryocytes (white) and neutrophils (black). Clec-2 expression entirely overlapped with CD41 expression, but not with Ela2. (B) Ventral views of Clec1bfl/fl (wild-type), Clec1bΔPLT, and Clec1b−/− fetal lungs at E17.5. (C) Neonatal lethality of Clec1bfl/fl and Clec1bΔPLT neonates. *P = .00127, Fisher’s exact test. (D) Immunostaining of α-SMA (green) and DAPI (blue) in E17.5 distal lung. AL, alveolar sac; M, lung mesothelium. (E) Quantification of α-SMA expression in Clec1bfl/fl and Clec1bΔPLT distal lung at E17.5; mean ± SD, n = 3 each. *P = .0211; **P = .0033, Holm-Sidak test. (F) Whole-mount immunohistochemistry of Wt1 (magenta) and Pdpn (green) in E17.5 luMCs. Arrowheads: Wt1-negative luMCs. (G) Quantification of luMC density; mean ± SD, n = 3 each. *P = .0281, Student t test. (H) Quantification of Wt1-negative rate in luMCs; mean ± SD, n = 3 each. N. S., not significant; P = .062, Student t test. (I) Platelet counts in E17.5 Clec1b+/+, Clec1b−/−, Clec1bfl/fl, and Clec1bΔPLT fetal blood; mean ± SD, n = 6, 6, 3, and 4, respectively. N.S., not significant, Tukey’s test. Scale bars: 25 μm (A,D,F); 1 mm (B).
Accordingly, Clec-2 expression was detected at a low level in Clec1bΔPLT fetal platelets; it was approximately 2% of the expression in Clec1bfl/fl fetal platelets (Figure 5A; supplemental Figure 3). We hypothesized that complete depletion of the remaining Clec-2 from platelets or thrombocytopenia in Clec1bΔPLT fetuses would enhance the lung phenotypic severity. Previous studies have shown that anti-Clec-2 antibody administration in mice specifically depleted Clec-2 on platelets,13,31 and that an anti-GPIb antibody induced continuous thrombocytopenia.32 We confirmed that nonmurine IgG intraperitoneally injected into the pregnant mice was transported to fetal blood (supplemental Figure 4) and that injection of an anti-Clec-2 antibody or anti-GPIb antibody to pregnant mice carrying Clec1bΔPLT fetuses induced complete depletion of Clec-2 in fetal Clec1bΔPLT platelets (supplemental Figure 3) without reducing fetal platelet counts (Figure 5B), or approximately 90% reduction in fetal platelet number (Figure 5B), respectively. Clec1bΔPLT fetuses treated with each antibody showed severe phenotypic characteristics that were nearly identical to those observed in Clec1b−/− fetuses: high neonatal lethality (Figure 5C), impaired α-SMA expression in distal lung interstitium (Figure 5D-E), and low Wt1-negative rate and high luMC density (Figure 5F-H). In 6 experimental sets of mice showing more than 10% neonatal lethality, Clec-2 expression levels in fetal platelets strongly correlated with the neonatal lethality rate (Figure 5I). These results demonstrate that Clec-2 expressed on platelets plays an essential role in normal lung development and neonatal survival.
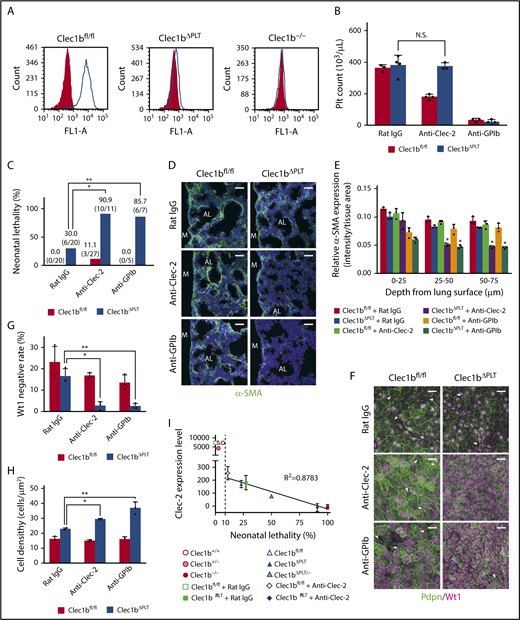
Platelets are required for lung development. (A) Clec-2 expression on platelets in E17.5 Clec1bfl/fl (wild-type), Clec1bΔPLT and Clec1b−/− fetuses. Gray-filled and open histograms indicate isotype control and Clec-2, respectively. (B) Platelet counts at E17.5 in Clec1bfl/fl and Clec1bΔPLT fetal blood treated with rat IgG (control), anti-Clec-2 antibody, or anti-GPIb antibody. Mean ± SD, n = 3, 4, 4, 3, 3, and 4, respectively, from the left of the histogram. N.S., not significant, Tukey’s test. (C) Neonatal lethality in Clec1bfl/fl and Clec1bΔPLT neonates treated with rat IgG (control), anti-Clec-2, or anti-GPIb. *P = .0021, **P = .0237, Fisher’s exact test. (D) Immunostaining of α-SMA (green) and DAPI (blue) in E17.5 distal lung. AL, alveolar sac; M, lung mesothelium. (E) Quantification of α-SMA expression in E17.5 distal lung. Asterisks indicate the 2 groups in which α-SMA expression was significantly lower than that in the other 4 groups; mean ± SD, n = 3 each; *P < .005, Holm-Sidak test. (F) Whole-mount immunohistochemistry of Wt1 (magenta) and Pdpn (green) in E17.5 luMCs. Arrowheads: Wt1-negative luMCs. (G) Quantification of Wt1-negative rate in luMCs; mean ± SD, n = 3 each. *P = .0019; **P = .0022, Tukey’s test. (H) Quantification of luMC density; mean ± SD, n = 3 each. *P = .0034; **P < .0001, Tukey’s test. (I) Correlation between Clec-2 expression in E17.5 fetal platelets (supplemental Figure 3) and neonatal lethality in the listed groups (Figures 1A, 4C, and 5C and Clec1bΔPLT/−); mean ± SD of Clec-2 expression level is shown. Numbers of individuals in all groups are shown in supplemental Figure 3. A very strong correlation (R2 = 0.8783) was observed in the 6 experimental sets of mice exhibiting more than 10% neonatal lethality (right side of the dashed line). Neonatal lethality of Clec1bΔPLT/− was 50.0% (5/10). Scale bars: 25 μm (D,F).
Platelets are required for lung development. (A) Clec-2 expression on platelets in E17.5 Clec1bfl/fl (wild-type), Clec1bΔPLT and Clec1b−/− fetuses. Gray-filled and open histograms indicate isotype control and Clec-2, respectively. (B) Platelet counts at E17.5 in Clec1bfl/fl and Clec1bΔPLT fetal blood treated with rat IgG (control), anti-Clec-2 antibody, or anti-GPIb antibody. Mean ± SD, n = 3, 4, 4, 3, 3, and 4, respectively, from the left of the histogram. N.S., not significant, Tukey’s test. (C) Neonatal lethality in Clec1bfl/fl and Clec1bΔPLT neonates treated with rat IgG (control), anti-Clec-2, or anti-GPIb. *P = .0021, **P = .0237, Fisher’s exact test. (D) Immunostaining of α-SMA (green) and DAPI (blue) in E17.5 distal lung. AL, alveolar sac; M, lung mesothelium. (E) Quantification of α-SMA expression in E17.5 distal lung. Asterisks indicate the 2 groups in which α-SMA expression was significantly lower than that in the other 4 groups; mean ± SD, n = 3 each; *P < .005, Holm-Sidak test. (F) Whole-mount immunohistochemistry of Wt1 (magenta) and Pdpn (green) in E17.5 luMCs. Arrowheads: Wt1-negative luMCs. (G) Quantification of Wt1-negative rate in luMCs; mean ± SD, n = 3 each. *P = .0019; **P = .0022, Tukey’s test. (H) Quantification of luMC density; mean ± SD, n = 3 each. *P = .0034; **P < .0001, Tukey’s test. (I) Correlation between Clec-2 expression in E17.5 fetal platelets (supplemental Figure 3) and neonatal lethality in the listed groups (Figures 1A, 4C, and 5C and Clec1bΔPLT/−); mean ± SD of Clec-2 expression level is shown. Numbers of individuals in all groups are shown in supplemental Figure 3. A very strong correlation (R2 = 0.8783) was observed in the 6 experimental sets of mice exhibiting more than 10% neonatal lethality (right side of the dashed line). Neonatal lethality of Clec1bΔPLT/− was 50.0% (5/10). Scale bars: 25 μm (D,F).
Pdpn on LECs is the partner of Clec-2 on platelets in lung development
Pdpn−/− mice reportedly show neonatal lethality resulting from respiratory failure; however, the definitive cause of the phenotype has not been established.33,34 We found that Pdpn−/− mice exhibited Clec1b−/−-like lung developmental abnormalities (supplemental Figure 5). The similar phenotypes strongly suggested that an interaction between Clec-2 and Pdpn is involved in lung development. Pdpn is widely used as a marker of AECs; however, it is unclear whether Pdpn is expressed in other tissues in the developing lung. Immunohistological analysis revealed Pdpn expression in AECs, luMCs, and LECs in wild-type lung parenchyma at E17.5 (Figure 6A; Pdpnfl/fl). Next, to identify the Pdpn-expressing cells required for lung development, we generated 3 tissue-specific Pdpn-deficient mouse lines by crossing Pdpnfl/fl mice with Shh-EGFPCre (AECs), Wt1-EGFPCre (luMCs), and Tie2-Cre (LECs) mice, with tissue-specific Pdpn deletion confirmed by immunostaining in each line (Figure 6A). The Tie2-Cre, Pdpnfl/fl mice showed high neonatal lethality (Figure 6B) and lung developmental abnormalities, including lumpy lung surface, impaired α-SMA expression in distal lung interstitium, and abnormal Wt1 expression pattern (Figure 6C-F). Conversely, Shh-EGFPCre, Pdpnfl/fl and Wt1-EGFPCre, Pdpnfl/fl mice did not show neonatal lethality or any lung developmental abnormalities (Figure 6B-F). Genomic recombination by Tie2-Cre transgenic mice used in this study was reportedly observed not only in LECs but also in vascular endothelial cells and blood cells.35 However, Pdpn was not detected in both vascular endothelial cells and blood cells in wild-type lungs (supplemental Figure 6). Given that both Clec-2 and Pdpn are membrane proteins, these results suggested that direct interaction between Clec-2 on platelets and Pdpn on LECs is involved in normal lung development.
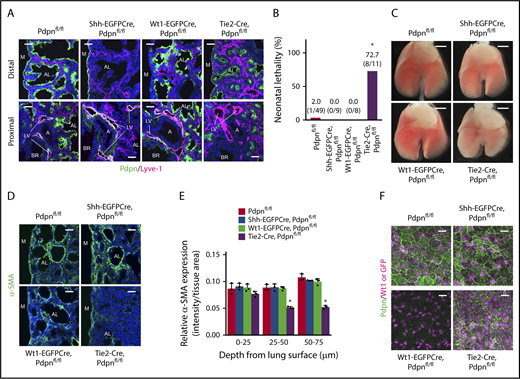
Pdpn on LECs partners with platelet Clec-2 for lung development. (A) Pdpn expression (green) in wild-type and AEC-, luMC-, and LEC-specific Pdpn-deficient lungs (Shh-EGFPCre, Wt1-EGFPCre, and Tie2-Cre, respectively). Lyve-1 (magenta) and DAPI (blue) were co-stained with Pdpn to visualize lymphatic vessels and nuclei. (B) Neonatal lethality in tissue-specific Pdpn-deficient mice. *P = .0000, Fisher’s exact test. (C) Ventral views of E17.5 whole lung. (D) Immunostaining of α-SMA (green) and DAPI (blue) in E17.5 distal lung. (E) Quantification of α-SMA expression in E17.5 distal lung. The asterisk indicates the group in which α-SMA expression was significantly lower than that in the other 3 groups; mean ± SD, n = 3 each. *P < .0001, Holm-Sidak test. (F) Whole-mount immunohistochemistry of Wt1 (magenta) or GFP (magenta) and Pdpn (green) in luMCs. GFP staining was performed only in Wt1-EGFPCre, Pdpnfl/fl luMCs because this strain is heterozygous for the Wt1 gene, and therefore the intensity of Wt1 staining in these mice was indistinct from that in other strains. Pdpn was not detected in Wt1-EGFPCre, Pdpnfl/fl luMCs, as Pdpn was deleted by Wt1-EGFPCre. A, artery; AL, alveolar sac; BR, bronchiole; LV, lymphatic vessel; M, lung mesothelium. Scale bars: 25 μm (A, D, and F); 1 mm (C).
Pdpn on LECs partners with platelet Clec-2 for lung development. (A) Pdpn expression (green) in wild-type and AEC-, luMC-, and LEC-specific Pdpn-deficient lungs (Shh-EGFPCre, Wt1-EGFPCre, and Tie2-Cre, respectively). Lyve-1 (magenta) and DAPI (blue) were co-stained with Pdpn to visualize lymphatic vessels and nuclei. (B) Neonatal lethality in tissue-specific Pdpn-deficient mice. *P = .0000, Fisher’s exact test. (C) Ventral views of E17.5 whole lung. (D) Immunostaining of α-SMA (green) and DAPI (blue) in E17.5 distal lung. (E) Quantification of α-SMA expression in E17.5 distal lung. The asterisk indicates the group in which α-SMA expression was significantly lower than that in the other 3 groups; mean ± SD, n = 3 each. *P < .0001, Holm-Sidak test. (F) Whole-mount immunohistochemistry of Wt1 (magenta) or GFP (magenta) and Pdpn (green) in luMCs. GFP staining was performed only in Wt1-EGFPCre, Pdpnfl/fl luMCs because this strain is heterozygous for the Wt1 gene, and therefore the intensity of Wt1 staining in these mice was indistinct from that in other strains. Pdpn was not detected in Wt1-EGFPCre, Pdpnfl/fl luMCs, as Pdpn was deleted by Wt1-EGFPCre. A, artery; AL, alveolar sac; BR, bronchiole; LV, lymphatic vessel; M, lung mesothelium. Scale bars: 25 μm (A, D, and F); 1 mm (C).
TGF-β signaling mediates Clec-2/Pdpn-dependent lung development
Next, we examined how the interaction between platelet Clec-2 and LEC Pdpn regulates luMC and adMYF differentiation. Syk is an essential molecule downstream of Clec-2 on platelet activation.36 We found that Syk−/− mice showed neonatal lethality and Clec1b−/−-like lung developmental abnormalities (supplemental Figure 7). In contrast, mice lacking the Pdpn cytoplasmic tail are viable.37 In addition, Clec-2 Y7A knock-in mice that maintain Clec-2 expression but cannot activate platelets show neonatal lethality.38 These reports, together with our results in Syk−/− mice, suggest that signal transduction within platelets is more critical than that within LECs.
Platelets contain various growth factors, including TGF-β, PDGF, and VEGF in α-granules,17,18 with TGF-β, in particular, being reported to promote myofibroblastic differentiation in mesothelial cells.39-41 Thus, we evaluated the effects of activated-platelet supernatant (APS), which contains platelet granule contents, on myofibroblastic differentiation in in vitro mesothelial cell (CCL-216) cultures and ex vivo lung explant cultures. As acidification by HCl forcedly converts the latent TGF-β to the active form,42 we used acidified-APS (APS-HCl) to enhance the effect of TGF-β in both experiments. APS-HCl has strong promyofibroblastic, antiproliferative, and antimesothelial effects on luMCs in vitro (supplemental Figure 8A-E). In lung explant experiments, APS-HCl also strongly induced myofibroblast differentiation in the interstitium of the primary septa (supplemental Figure 8F). APS had similar but only minor effects compared with APS-HCl (supplemental Figure 8A-F). Both the in vitro and ex vivo results suggested that TGF-β is a signaling molecule that regulates adMYF differentiation of luMCs in platelet Clec-2-dependent lung development.
To evaluate the requirement for TGF-β in Clec-2/Pdpn-dependent lung development in vivo, we analyzed the phenotype of Clec1bfl/fl and Clec1bΔPLT fetuses treated with an anti-TGF-β neutralization antibody. Although anti-TGF-β antibody treatment had no effect on wild-type (Clec1bfl/fl), the treatment enhanced phenotypic severity in Clec1bΔPLT fetuses to a level comparable to that in Clec1b−/− fetuses, the phenotype included high neonatal lethality (Figure 7A), impaired α-SMA expression in distal lung interstitium (Figure 7B-C), low Wt1-negative rate, and high luMC density (Figure 7D-F) without reduction of platelet Clec-2 expression (Figure 7G). More than 99% of TGF-β in both Clec1b+/+ and Clec1b−/− fetal platelets was TGF-β1 (supplemental Figure 8G); importantly, a significant amount of TGF-β1 in the developing lung was derived from platelets (supplemental Figure 8H), and TGF-β1 concentration in the developing lung was significantly lower in Clec1b−/− than in Clec1b+/+ mice (Figure 7H). These results suggest that TGF-β1, which is released from platelets and/or produced in lung parenchymal cells by stimulation of platelet granule contents, plays a crucial role in adMYF differentiation of luMCs in Clec-2/Pdpn-dependent lung development.
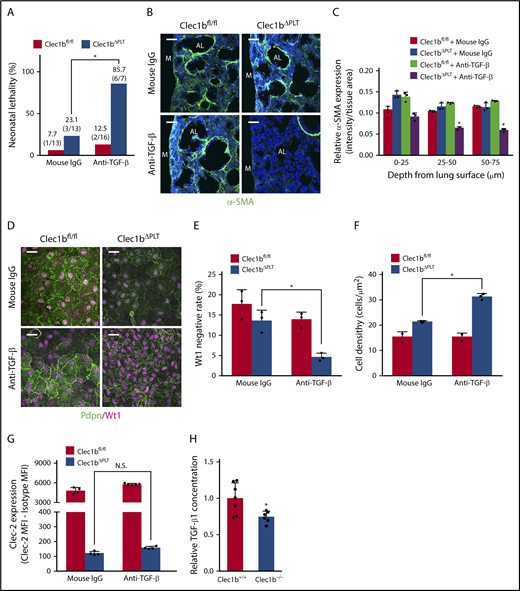
TGF-β mediates Clec-2-Pdpn-dependent lung development. (A) Neonatal lethality in Clec1bfl/fl and Clec1bΔPLT neonates treated with mouse IgG (control) or anti-TGF-β. *P = .0049, Fisher’s exact test. (B) Immunostaining of α-SMA (green) and DAPI (blue) in E17.5 distal lung. AL, alveolar sac; M, lung mesothelium. (C) Quantification of α-SMA expression in E17.5 distal lung. The asterisk indicates the group in which α-SMA expression was significantly lower than that in the other 3 groups; mean ± SD, n = 3 each. *P < .0001, Holm-Sidak test. (D) Whole-mount immunohistochemistry of Wt1 (magenta) and Pdpn (green) in E17.5 luMCs. (E) Quantification of Wt1-negative rate in luMCs; mean ± SD, n = 3 each. *P = .0084, Tukey’s test. (F) Quantification of luMC density; mean ± SD, n = 3 each. *P = .0001, Tukey’s test. (G) Clec-2 expression on fetal platelets in fetuses treated with anti-TGF-β antibody; mean ± SD, n = 4, 4, 5, and 4, respectively, from the left of the histogram; Student t test. N.S., not significant. (H) Total TGF-β1 relative concentration in Clec1b+/+ and Clec1b–/– lungs; mean ± SD, n = 7 (Clec1b+/+), n = 6 (Clec1b–/–). *P = .0187, Student t test. Scale bars: 25 μm (B,D).
TGF-β mediates Clec-2-Pdpn-dependent lung development. (A) Neonatal lethality in Clec1bfl/fl and Clec1bΔPLT neonates treated with mouse IgG (control) or anti-TGF-β. *P = .0049, Fisher’s exact test. (B) Immunostaining of α-SMA (green) and DAPI (blue) in E17.5 distal lung. AL, alveolar sac; M, lung mesothelium. (C) Quantification of α-SMA expression in E17.5 distal lung. The asterisk indicates the group in which α-SMA expression was significantly lower than that in the other 3 groups; mean ± SD, n = 3 each. *P < .0001, Holm-Sidak test. (D) Whole-mount immunohistochemistry of Wt1 (magenta) and Pdpn (green) in E17.5 luMCs. (E) Quantification of Wt1-negative rate in luMCs; mean ± SD, n = 3 each. *P = .0084, Tukey’s test. (F) Quantification of luMC density; mean ± SD, n = 3 each. *P = .0001, Tukey’s test. (G) Clec-2 expression on fetal platelets in fetuses treated with anti-TGF-β antibody; mean ± SD, n = 4, 4, 5, and 4, respectively, from the left of the histogram; Student t test. N.S., not significant. (H) Total TGF-β1 relative concentration in Clec1b+/+ and Clec1b–/– lungs; mean ± SD, n = 7 (Clec1b+/+), n = 6 (Clec1b–/–). *P = .0187, Student t test. Scale bars: 25 μm (B,D).
Discussion
Here, we report that platelet activation via a Clec-2/Pdpn pathway plays an essential role in luMC and adMYF differentiation and subsequent primary septum formation during lung development. We also identify LECs as Pdpn-expressing cells partnering with platelets via Clec-2, and TGF-β as a downstream signaling molecule in the Clec-2/Pdpn-dependent lung developmental process.
In Clec1b−/− mice, abnormal lung morphology was observed from E17.5, when the morphology of distal lung parenchyma drastically changes. In this period, adMYFs were first observed in the interstitium between alveolar sacs in Clec1b+/+ fetuses, but very few adMYFs were observed in Clec1b−/− fetuses. In general, myofibroblasts are contractile and secrete ECM, and it has been reported that impaired adMYF differentiation is accompanied by decreased elastogenesis.4,5,9 Taken together, these findings indicate that the thick primary septa and subsequent decreased elastogenesis in the Clec1b−/− lung are caused by a lack of normal adMYFs.
Lineage-tracing analysis using Wt1-EGFPCre, CAG-CAT-EGFP mice showed that a majority of adMYFs are luMC-descendant cells. Inspired by this observation, we aimed to unravel the molecular mechanism underlying luMC differentiation into adMYFs. We considered Wt1 to be a negative regulator of adMYF differentiation of luMCs. Wt1 knockdown by shRNA or TGF-β1 treatment induces expression of mesothelial-to-mesenchymal transition markers and α-SMA in human luMCs.39,41 In our observations in vivo, Wt1 expression disappeared in a portion of luMCs and adMYFs differentiated normally in Clec1b+/+ lungs. In addition, APS-HCl had promyofibroblastic, antiproliferative, and antimesothelial effects on CCL-216 cells. We also found hyperproliferation and high density with abnormally maintained Wt1 expression in Clec1b−/− luMCs. Furthermore, we showed that TGF-β neutralization in vivo caused abnormally maintained Wt1 expression, high density of luMCs, and impaired adMYF differentiation. Hence, we propose that appropriate Wt1 downregulation by TGF-β is important for luMC proliferation and adMYF differentiation during normal lung development. Thus, decreased TGF-β1 by Clec-2 deletion may inhibit the differentiation of mesothelial cell-derived adMYFs, although we cannot exclude the possibility that decreased TGF-β1 also inhibits the differentiation of nonmesothelial cell-derived adMYFs. In contrast, previous reports have revealed that other signaling pathways such as Pdgfa, Notch, and glucocorticoid pathways are involved in the process from luMC differentiation to alveolar elastogenesis.4,5,9 Integrated understanding of the complex interaction among multiple signaling pathways including Clec-2/Pdpn is required in the future.
The lung developmental process has been generally explained by interactions among lung tissues. Our findings reveal for the first time that platelets are indispensable players in normal lung development. Certain knockout mouse lines exhibit thrombocytopenia, but impaired lung development has not been described in these mice. For example, Nfe2-deficient mice exhibit low numbers of abnormal platelets (50–100 × 103/μL),43,44 but the mice present normal appearance and histology of major organs.43 In our study, anti-GPIb antibody-treated wild-type (Clec1bfl/fl) fetuses showed decreased platelet number (<40 × 103/μL), but no lung developmental abnormalities or neonatal lethality. These findings suggest that lung developmental abnormalities caused by thrombocytopenia require a further reduction in platelet number or a combination of thrombocytopenia and specific qualitative platelet abnormalities, such as a marked reduction in Clec-2 expression, as observed in Clec1bΔPLT mice. Alternatively, similar to Clec1bΔPLT, Nfe2-deficient mice may exhibit minor lung developmental abnormalities.
The Clec1b−/−-like phenotypes in Syk−/− mice and the effects of APS or APS-HCl in in vitro and ex vivo experiments suggest that granule contents released from platelets on Clec-2-dependent platelet activation are involved in lung development. TGF-β neutralization by antibody treatment enhanced the phenotypic severity in Clec1bΔPLT fetuses, without decreasing Clec-2 expression on platelets. In addition, nearly all TGF-β in platelets was TGF-β1, and the amount of TGF-β1 in Clec1b−/− lungs was significantly lower than that in Clec1b+/+ lungs. Collectively, it is suggested that TGF-β1 acts downstream of Clec-2 in lung development. However, no report has been published to date, showing lung developmental abnormalities in Tgfb1−/− mice.45 It is possible that other platelet-released molecules, such as PDGF-A, which is a well-known positive regulator of adMYF differentiation, have redundant or synergistic roles with TGF-β in lung development. In Clec1bΔPLT fetuses, the amounts of all molecules released from platelets by Clec-2-dependent platelet activation are predicted to be considerably lower than those in wild-type fetuses. This, combined with TGF-β neutralization, might have caused the Clec1b−/−-like phenotype in these mice.
Pdpn expression is detected in AECs, luMCs, and LECs, but not in vascular endothelial cells and blood cells, during lung development. Pdpn−/− mice show lung developmental abnormalities and respiratory failure; however, the Pdpn-expressing cell types responsible for normal lung development remain to be defined. We found that only Tie2-Cre, Pdpnfl/fl fetuses exhibited Clec1b−/−-like lung developmental abnormalities among 3 tissue-specific Pdpn-knockout mice. This observation strongly suggested that Pdpn on LECs, not AECs and luMCs, partners with Clec-2 on platelets for lung development. Previous reports demonstrated that LECs activate platelets through Clec-2/Pdpn binding.14,46 Thus, we proposed that direct interaction between platelet Clec-2 and LEC Pdpn leads to TGF-β1 release through platelet activation, which results in normal differentiation of luMCs and adMYFs. As we used several Cre-loxP systems and antibodies to prove our hypothesis, notably, the limitations of this study include the specificity of Cre expression systems and that of antibodies.
The sites in which platelets might directly contact LECs to promote lung development remain unclear because blood generally does not flow in lymphatic vessels. The interaction might occur at the lymphovenous junction between the subclavian vein and the thoracic duct.47 Alternatively, blood and lymphatic capillaries might make transient contacts in the lungs and/or entire body. We and others have previously reported misconnections between peripheral blood and lymphatic capillaries in mice lacking any molecule in the Clec-2/Pdpn pathway.22,48,49 This implies that developing blood and lymphatic capillaries are in direct contact, and that platelet Clec-2 frequently interacts with LEC Pdpn.
In idiopathic pulmonary fibrosis (IPF), lung tissue becomes thickened and stiff through deposition of ECM from excessively differentiated and proliferated α-SMA+ myofibroblasts, and TGF-β is closely involved in the exacerbation of IPF.50,51 In contrast, α-SMA-positive cells that newly appear in the lung mesothelium and submesothelial area were recently reported to contribute to neoalveolarization in compensatory growth after pneumonectomy.52,53 Our findings support the novel concept that platelets participate in the development of solid organs and provide clinical implications that platelets might participate in pathophysiological situations related to lung myofibroblast differentiation in adults, such as IPF and neoalveolarization after pneumonectomy in patients with IPF or cancer.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Hisaichiro Nakazawa, Uina Fukuda, Junko Nakagomi, and Takako Otsuka (University of Yamanashi) for technical assistance, and Yuki Asano (The Jikei University School of Medicine) for transmission electron microscopy analysis. This study was supported in part by KAKENHI (16K19572) and the Funding Program for Next Generation World-leading Researchers (LS052).
Authorship
Contribution: N. Tsukiji designed the study; N. Tsukiji, O.I., N. Tatsumi, H.N., K.U., T. Shirai, T. Sasaki, S.O., S.T., and T.T. performed experiments; M.M., N. Tatsumi, M.O., and M.H. provided mouse strains essential for this study; M.M., M.H., and Y.O. provided important insight and advice; and N. Tsukiji and K.S.-I. were responsible for assembling figures and writing the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
The current affiliation for T. Shirai is the Department of Biomedical Engineering, Oregon Health & Science University, Portland, OR.
The current affiliation for S. Tamura is the Department of Pathophysiological Laboratory Sciences, Nagoya University Graduate School of Medicine, Nagoya, Aichi, Japan.
Correspondence: Katsue Suzuki-Inoue, Department of Clinical and Laboratory Medicine, Faculty of Medicine, University of Yamanashi, 1110 Shimokato, Chuo, Yamanashi 409-3898, Japan; e-mail: katsuei@yamanashi.ac.jp.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal