

Key Points
AMPK-ACC signaling in platelets is a key mechanism regulating primary hemostasis and arterial thrombosis.
AMPK-ACC signaling controls collagen-induced TXA2 generation and dense granule release by modulating platelet phospholipid content.

Abstract
AMP-activated protein kinase (AMPK) α1 is activated in platelets on thrombin or collagen stimulation, and as a consequence, phosphorylates and inhibits acetyl-CoA carboxylase (ACC). Because ACC is crucial for the synthesis of fatty acids, which are essential for platelet activation, we hypothesized that this enzyme plays a central regulatory role in platelet function. To investigate this, we used a double knock-in (DKI) mouse model in which the AMPK phosphorylation sites Ser79 on ACC1 and Ser212 on ACC2 were mutated to prevent AMPK signaling to ACC. Suppression of ACC phosphorylation promoted injury-induced arterial thrombosis in vivo and enhanced thrombus growth ex vivo on collagen-coated surfaces under flow. After collagen stimulation, loss of AMPK-ACC signaling was associated with amplified thromboxane generation and dense granule secretion. ACC DKI platelets had increased arachidonic acid-containing phosphatidylethanolamine plasmalogen lipids. In conclusion, AMPK-ACC signaling is coupled to the control of thrombosis by specifically modulating thromboxane and granule release in response to collagen. It appears to achieve this by increasing platelet phospholipid content required for the generation of arachidonic acid, a key mediator of platelet activation.
Introduction
A growing body of evidence shows that lipids are essential in regulating platelet functions. Indeed, the platelet exhibits a complex array of more than 5000 distinct lipid species, with more than 700 responding to thrombin activation.1 Given this increased focus on the role of lipid species in platelet function, it is imperative to understand the molecular basis of their regulation during platelet activation. Acetyl-CoA carboxylase (ACC) is a good candidate because of its established role as a central regulator of fatty acid metabolism.2 ACC catalyzes acetyl-CoA carboxylation to form malonyl-CoA. Its 2 isoforms, ACC1 and ACC2, have distinct cellular distributions.3 ACC1 is present in the cytosol and synthesizes malonyl-CoA for de novo lipogenesis,4 whereas ACC2 is localized on the outer mitochondrial membrane and generates malonyl-CoA, which inhibits fatty acid transport into mitochondria for oxidation.5,6 ACC is a bona fide substrate of AMP-activated protein kinase (AMPK), and its phosphorylation is typically used as a marker of AMPK activation in cells and tissues, including platelets.7-9 AMPK phosphorylates ACC1/2 on serine residues (Ser79/212), leading to suppression of ACC activity.10,11 We have previously reported that AMPKα1 is activated in platelets upon thrombin stimulation, and increases the phosphorylation of myosin regulatory light chains, cofilin, and vasodilator-stimulated phosphoprotein.7 These cytoskeletal proteins are critical for triggering platelet shape change and the centralization of secretory granules during platelet activation.12 However, the effect of AMPK-mediated ACC phosphorylation on platelet function has never been investigated.
Fatty acids fulfill at least 3 main roles: structural, signaling, and energy storage. Phospholipids (PL) are the major structural lipids in platelets. Upon platelet activation, reorganization of the plasma membrane PL facilitates shape change and filopodia and lamellipodia formation,13 as well as granule secretion14 or microvesicle formation. In addition to this structural role, PL provide substrates for phospholipases A (PLA) or C (PLC) to generate bioactive species, including phosphatidylinositides, 1,2-diacylglycerol (DAG), inositol 1,4,5-trisphosphate, and eicosanoids/prostaglandins.1 These secondary mediators are crucial for the tight regulation of platelet activation.15-17
Finally, lipids contribute to platelet energy metabolism. In the basal state, platelets are more oxidative than glycolytic,18,19 and lipid oxidation contributes to at least one-third of the total oxygen consumed by mitochondria.18,20,21 Mitochondrial lipid oxidation increases on thrombin stimulation to cope with the energetic demands of platelet activation,20,22 a process that is supported by the enhanced availability of eicosanoids released from the membrane PL through the action of Ca2+-dependent cytosolic phospholipase A2 (cPLA2).1
Clearly, the signaling needed to coordinate these diverse functions of PL metabolism is important to define. In the current study, we hypothesized that ACC controls key aspects of platelet function using mice with alanine knock-in mutations in both ACC1 (at Ser79) and ACC2 (at Ser212; ACC double knock-in [ACC DKI] mice). These mice harbor functional ACC and AMPK, but express a mutant form of ACC that can no longer be inhibited by AMPK phosphorylation, resulting in a persistent active form of the enzyme.2
Here, we report that blood from ACC DKI mice have increased thrombus formation on collagen-coated surfaces or after vascular injury. The underlying mechanisms involve elevated levels of arachidonic acid (AA)-containing phosphatidylethanolamine plasmalogen (PEP) lipids in ACC DKI platelets compared with wild-type (WT), increasing thromboxane A2 (TXA2) generation and granule secretion after platelet stimulation with collagen. Although mitochondrial fatty acid oxidation is important in thrombin-dependent platelet activation, we found that AMPK-ACC signaling had no effect on platelet bioenergetics.
These findings highlight a novel metabolic regulatory pathway in platelets that influences thrombus formation by modulating the content of specific PL, which generates key mediators of platelet activation.
Methods
Mice
ACC1/2 DKI mice have been described previously.2 WT mice served as controls. Eight- to 16-week-old male mice were studied. Animal procedures and protocols were approved by local authorities (Comité d’éthique facultaire pour l’expérimentation animale, 2012/UCL/MD/003 and 2016/UCL/MD/027) and performed in accordance with the Guide for the Care and Use of Laboratory Animals, published by the National Institutes of Health (NIH Publication No. 85-23, revised 1996).
Platelet preparation
Mice were bled under ketamine and xylazine anesthesia from the retro-orbital plexus. Blood was collected in 1/6 citrate-dextrose solution with apyrase 1 U/mL. Platelet-rich plasma was obtained by centrifugation at 800g for 5 seconds, followed by 5 minutes at 100g. It was washed by adding 2 volumes of citrate-dextrose with apyrase 1 U/mL. The platelets were pelleted by centrifugation at 400g for 5 minutes and resuspended to a density of 2.5 × 105/µL (unless stated otherwise) in modified Tyrode’s buffer. Platelets were counted with Cell-Dyn Emerald. They were stimulated with agonists in the presence of 2 mM CaCl2.
Flow chamber assay
Blood was collected from the retro-orbital plexus into 48 µM D-Phenylalanyl-prolyl-arginyl Chloromethyl Ketone, 5 U/mL heparin, and 40 U/mL fragmin. Samples of 400 µL were flowed over type I collagen-coated (100 µg/mL), von Willebrand factor (VWF)–binding protein (12.5 µg/mL) and laminin (50 µg/mL)-coated or VWF-binding protein (12.5 µg/mL), laminin (50 µg/mL), and rhodocytin (250 µg/mL)-coated coverslips mounted on a transparent, parallel plate flow chamber (50 µm depth, 3 mm width, and 20 mm length), at a shear rate of 1000 s−1 for 3.5 minutes, as described.23 Alternatively, samples were preincubated with 20 µM ticagrelor for 10 minutes or the corresponding vehicle (dimethyl sulfoxide) and flowed over type I collagen-coated coverslips, as described earlier. Activated platelets in thrombi were poststained with fluorescein isothiocyanate-labeled anti-P-selectin antibody (Ab; 1:40), PE-labeled JON/A Ab against the active conformation of αIIbβ3 (1:20), or Alexa Fluor 647-annexin A5 (1:200), all diluted in Tyrode’s buffer. Labeling was undertaken for 2 minutes (stasis), after which unbound Abs were removed by perfusion with Tyrode’s buffer. Brightfield phase-contrast and fluorescence images were recorded by a nonconfocal 2-camera system. Surface coverage was analyzed by ImagePro software (Media Cybernetics).24
Ferric chloride-induced thrombosis
Carotid arteries were injured in anesthetized mice by topical application of 10% ferric chloride (FeCl3) for 5 minutes, as described previously.25 Briefly, exogenous carboxyfluorescein succinimidyl ester-labeled platelets were injected into the jugular vein of anesthetized mice, and fluorescence was recorded by BX61WI microscope (Olympus). Digital images were captured every 2 minutes for a total of 24 minutes.
Untargeted lipidomics
Lipidomics analysis was carried out on the commercial Lipidyzer platform, according to the manufacturer’s instructions (Sciex).26 The amount of platelets needed to obtain consistent results was evaluated before application of the commercial platform. Lipid analysis was performed in flow-injection mode, separating lipid classes by differential mobility spectroscopy,26 followed by tandem mass spectrometry of lipid species with QTrap 5500 in multiple reaction monitoring mode. Lipid species were identified and quantified on the basis of characteristic mass spectrometric transitions. Commercial Lipidyzer software automatically calculated lipid species concentrations. All samples were analyzed in a randomized fashion. Control plasma samples, as well as fortified plasma samples, were assessed daily as quality controls. Relative standard deviations of quality control samples were below 15% for all lipid classes, except for sphingomyelin, for which a relative standard deviation of 25% was noted.
A detailed description of the reagents and the methods is provided in supplemental Methods, available on the Blood Web site.
Results
Lack of AMPK-ACC phosphorylation does not affect AMPK signaling or cytoskeletal protein phosphorylation
ACC WT and ACC DKI mice have comparable erythrocyte, leukocyte, and platelet counts in whole blood (supplemental Table 1). Expression levels of the major platelet surface receptors, αIIbβ3, GPIbα, the collagen receptor GPVI, and the protease-activated receptor-3 and protease-activated receptor-4, were equivalent in ACC WT and ACC DKI platelets (supplemental Figure 1A-B).
ACC1, but not ACC2, was detected in both human and murine platelets (Figure 1A). Consistent with previous observations,7 0.2 U/mL thrombin (Figure 1B) or 5 µg/mL collagen (Figure 1C) induced phosphorylation on Ser79 of ACC in platelets from WT mice with maximal effects after 1 and 5 minutes, respectively. As expected, no Ser79 ACC phosphorylation was detected in either basal or thrombin- or collagen-treated ACC DKI platelets (Figure 1B-C). ACC1 mutation did not influence AMPK activation, reflected by Thr172 phosphorylation (Figure 1D-E). In addition, thrombin- or collagen-induced phosphorylation of known AMPK cytoskeletal targets, myosin light chain, cofilin, and vasodilator-stimulated phosphoprotein were similar between ACC DKI and ACC WT platelets (Figure 1D-E), as well as platelet-spreading and lamellipodia/filopodia formation after platelet immobilization on fibrinogen-coated coverslips (supplemental Figure 1C-H). These data confirm that although ACC phosphorylation was impaired, AMPK signaling was unaltered in ACC DKI platelets.
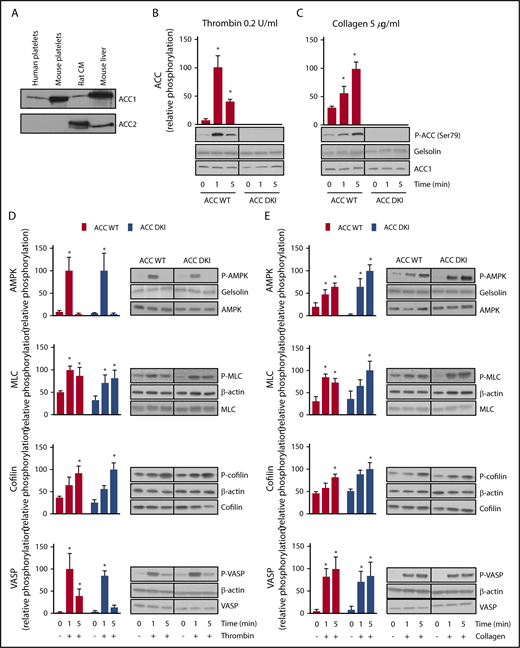
Lack of AMPK-ACC phosphorylation does not affect AMPK signaling or phosphorylation of cytoskeletal proteins. (A) Washed murine and human platelets were lysed and subjected to western blotting for ACC1 or ACC2 isoform expression analysis. Isolated rat cardiomyocytes (CM) and mouse liver extracts served as positive controls for the detection of ACC2 and ACC1, respectively. (B-E) ACC WT and ACC DKI platelets were stimulated with 0.2 U/mL thrombin (B,D) or 5 µg/mL collagen (C,E) for the indicated times. Whole-platelet lysates were subjected to western blotting and probed with Ser79 phosphorylated ACC Ab (B-C), Thr172 phosphorylated AMPK, Ser19 phosphorylated myosin light chain (MLC), Ser3 phosphorylated cofilin and Thr278 phosphorylated vasodilator-stimulated phosphoprotein (VASP) Abs (D-E). Gelsolin and β-actin were used as loading controls. Quantification and representative western blotting are systematically shown. The solid lines on the western blots indicate that samples were run on the same gel but were not contiguous. The results are expressed as means ± standard error of the mean (SEM; at least 3 experiments for each condition). *Values statistically different from respective untreated platelets; P ≤ .05. Analysis was performed by 2-way analysis of variance (ANOVA). See also supplemental Figure 1 and supplemental Table 1.
Lack of AMPK-ACC phosphorylation does not affect AMPK signaling or phosphorylation of cytoskeletal proteins. (A) Washed murine and human platelets were lysed and subjected to western blotting for ACC1 or ACC2 isoform expression analysis. Isolated rat cardiomyocytes (CM) and mouse liver extracts served as positive controls for the detection of ACC2 and ACC1, respectively. (B-E) ACC WT and ACC DKI platelets were stimulated with 0.2 U/mL thrombin (B,D) or 5 µg/mL collagen (C,E) for the indicated times. Whole-platelet lysates were subjected to western blotting and probed with Ser79 phosphorylated ACC Ab (B-C), Thr172 phosphorylated AMPK, Ser19 phosphorylated myosin light chain (MLC), Ser3 phosphorylated cofilin and Thr278 phosphorylated vasodilator-stimulated phosphoprotein (VASP) Abs (D-E). Gelsolin and β-actin were used as loading controls. Quantification and representative western blotting are systematically shown. The solid lines on the western blots indicate that samples were run on the same gel but were not contiguous. The results are expressed as means ± standard error of the mean (SEM; at least 3 experiments for each condition). *Values statistically different from respective untreated platelets; P ≤ .05. Analysis was performed by 2-way analysis of variance (ANOVA). See also supplemental Figure 1 and supplemental Table 1.
ACC DKI mice display enhanced primary hemostasis and thrombosis
The effect of AMPK-mediated ACC phosphorylation on hemostasis and thrombosis was evaluated in vivo. No spontaneous bleeding or thrombotic event was observed in 91 ACC DKI young mice (up to 4 months) or 19 ACC DKI older mice (at least 9 months old). However, median tail bleeding time was significantly shorter in ACC DKI mice than in their WT counterparts (ACC DKI: 82 ± 42 seconds vs ACC WT: 188 ± 51 seconds; P < .05; Figure 2A). We then investigated the role of ACC phosphorylation on arterial thrombosis in vivo, in the collagen-dependent carotid artery thrombosis model, in response to a 10% FeCl3 application. Arterial thrombus formation was monitored in real time by intravital fluorescence microscopy (Figure 2B-C). The rate of thrombus growth was significantly increased in ACC DKI mice compared with the WT animals. The fold change increase in thrombus growth during a 20-minute period after arterial injury was 62.4 ± 17.4 for ACC DKI mice vs 31.3 ± 8.2 for ACC WT mice relative to baseline (Figure 2B). These data demonstrate that AMPK-ACC signaling is a key mechanism regulating primary hemostasis and arterial thrombosis in vivo.
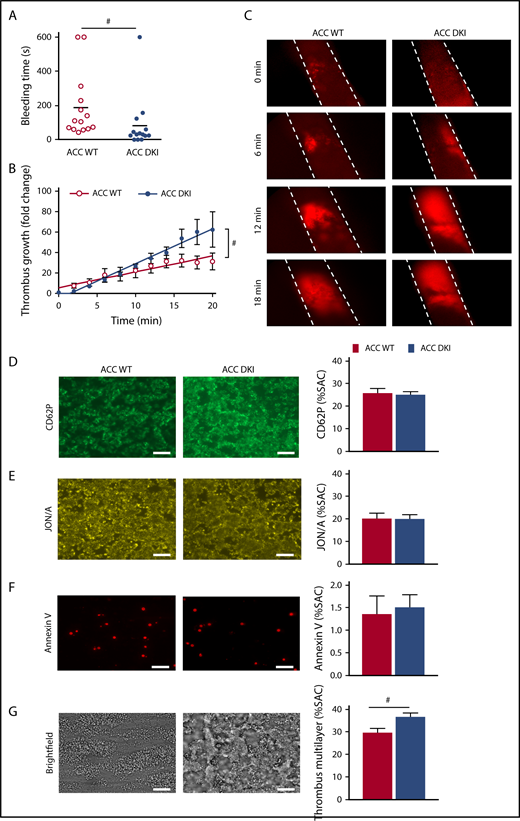
ACC DKI mice display enhanced primary hemostasis and thrombosis. (A) Tail bleeding times of ACC WT and ACC DKI mice in saline at 37°C. Individual values are plotted on the graph (n = 14 in each group). Bars indicate means. #P ≤ .05. Data were analyzed by the Mann-Whitney U test. (B-C) ACC WT and ACC DKI mice were subjected to in vivo FeCl3-induced thrombosis of carotid arteries (10% FeCl3, 5 min). Thrombus formation was monitored by analyzing exogenous carboxyfluorescein succinimidyl ester-labeled platelet accumulation by intravital microscopy and recording videos (fluorescence) of microscopic images every 2 minutes. (B) Thrombus growth kinetics was evaluated by dividing the area of the thrombus at time (t) by the area of the same thrombus at time 0, defined as the time at which the thrombus first reaches 100 µm. Thrombus growth is expressed as means ± SEM per group at different times with fitted regression lines (at least 6 mice/group). Slopes are statistically different on the basis of significant interaction (#P ≤ .05) between slope and group in a linear model, with group, time, and their interaction as covariates. (C) Representative fluorescence microscopy images at 0, 6, 12, and 18 minute after FeCl3 application (original magnification ×10). (D-G) Whole blood from ACC WT and ACC DKI mice was perfused over collagen-coated surfaces (100 µg/mL) at a shear rate of 1000 s−1. Exposure of P-selectin was evaluated by staining with CD62P Ab (D), αIIbβ3 integrin activation by JON/A Ab (E) and phosphatidylserine externalization by Annexin V (F). Thrombus formation was assessed on brightfield images taken 3.5 minutes after initial blood perfusion (G). Representative images appear on the left. Scale bars represent 20 µm. Histograms indicate quantification of surface area covered (SAC) by P-selectin (D), activated αIIbβ3 (E), Annexin V (F)–positive platelets or multilayered platelet thrombi (G). The results are expressed as means ± SEM (at least 4 mice/group). #P ≤ .05. Data were analyzed by the Mann-Whitney U test.
ACC DKI mice display enhanced primary hemostasis and thrombosis. (A) Tail bleeding times of ACC WT and ACC DKI mice in saline at 37°C. Individual values are plotted on the graph (n = 14 in each group). Bars indicate means. #P ≤ .05. Data were analyzed by the Mann-Whitney U test. (B-C) ACC WT and ACC DKI mice were subjected to in vivo FeCl3-induced thrombosis of carotid arteries (10% FeCl3, 5 min). Thrombus formation was monitored by analyzing exogenous carboxyfluorescein succinimidyl ester-labeled platelet accumulation by intravital microscopy and recording videos (fluorescence) of microscopic images every 2 minutes. (B) Thrombus growth kinetics was evaluated by dividing the area of the thrombus at time (t) by the area of the same thrombus at time 0, defined as the time at which the thrombus first reaches 100 µm. Thrombus growth is expressed as means ± SEM per group at different times with fitted regression lines (at least 6 mice/group). Slopes are statistically different on the basis of significant interaction (#P ≤ .05) between slope and group in a linear model, with group, time, and their interaction as covariates. (C) Representative fluorescence microscopy images at 0, 6, 12, and 18 minute after FeCl3 application (original magnification ×10). (D-G) Whole blood from ACC WT and ACC DKI mice was perfused over collagen-coated surfaces (100 µg/mL) at a shear rate of 1000 s−1. Exposure of P-selectin was evaluated by staining with CD62P Ab (D), αIIbβ3 integrin activation by JON/A Ab (E) and phosphatidylserine externalization by Annexin V (F). Thrombus formation was assessed on brightfield images taken 3.5 minutes after initial blood perfusion (G). Representative images appear on the left. Scale bars represent 20 µm. Histograms indicate quantification of surface area covered (SAC) by P-selectin (D), activated αIIbβ3 (E), Annexin V (F)–positive platelets or multilayered platelet thrombi (G). The results are expressed as means ± SEM (at least 4 mice/group). #P ≤ .05. Data were analyzed by the Mann-Whitney U test.
Lack of AMPK-ACC signaling favors thrombus formation during perfusion on collagen in flow conditions
We next examined thrombus formation ex vivo, using a flow chamber system. Whole blood from ACC WT and DKI mice was perfused at an intermediate shear rate of 1000 s−1, on 3 different coated surfaces containing collagen, laminin, or VWF-binding protein in the absence or presence of rhodocytin.24 Brightfield images were captured to assess overall platelet adhesion and thrombus formation. Platelet activation was simultaneously monitored by infusing fluorescently labeled anti-CD62P Ab as a marker of α-granule secretion, JON/A Ab to measure αIIbβ3 activation, and annexin V to analyze phosphatidylserine externalization, reflecting platelet procoagulant activity.
On collagen-coated coverslips, the surface area covered by adherent platelets and their activation state (Figure 2D-F) were similar between the 2 genotypes. However, buildup of multilayered platelet thrombi was increased with ACC DKI blood compared with WT (Figure 2G), indicating that AMPK-ACC signaling affects mechanisms involved in the secondary formation of platelet aggregates on collagen. Of note, no differences in platelet adhesion and activation processes were observed between ACC DKI and WT blood perfused on laminin and VWF-binding protein in the absence or presence of rhodocytin (data not shown).
ACC DKI platelets display increased dense granule secretion and thromboxane generation on collagen stimulation
To further decipher the mechanisms responsible for the gain-of-function phenotype of ACC DKI mice on thrombus formation, we studied the effect of a lack of AMPK-mediated ACC phosphorylation on washed platelet reactivity. First, we investigated the effect of thrombin or collagen on αIIbβ3 inside-out activation by analyzing platelet aggregation. ACC DKI platelets aggregated normally at low and high concentrations of thrombin or collagen (Figure 3A; supplemental Figure 2A). Accordingly, thrombin- or collagen-induced activation of αIIbβ3, detected by JON/A Ab, was normal (Figure 3B). Integrin αIIbβ3-mediated clot retraction consistently showed no differences between ACC WT and ACC DKI platelets after thrombin stimulation (supplemental Figure 2B-C).
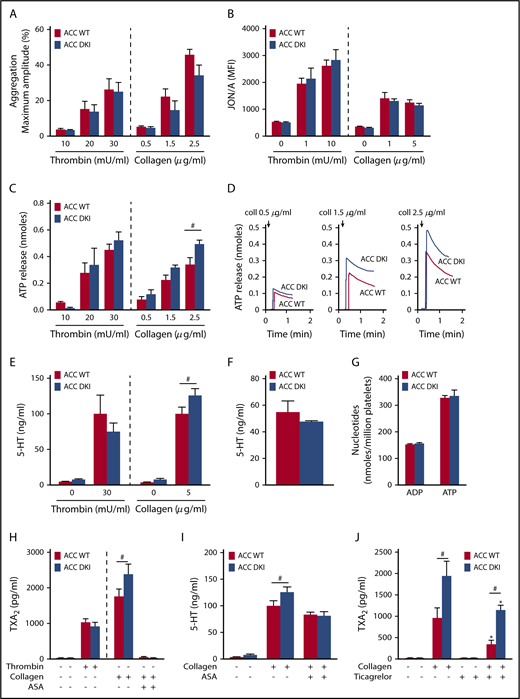
ACC DKI platelets display increased dense granule secretion and thromboxane generation on collagen stimulation. (A) Washed platelets were stimulated with thrombin or collagen at the indicated concentrations, and light transmission was measured (Chrono-Log). Aggregation is expressed as the maximal percentage of light transmitted. The dashed line represents separate analyses. The results are expressed as means ± SEM (at least 4 experiments for each condition). (B) αIIbβ3 activation (binding of JON/A) was analyzed by flow cytometry in washed ACC WT and DKI platelets stimulated with thrombin for 8 minutes or collagen for 30 minutes at the indicated concentrations. The dashed line represents separate analyses. The results are expressed as mean fluorescence intensity (MFI) ± SEM (at least 4 experiments for each condition). (C-D) Washed ACC WT and DKI platelets were stimulated with thrombin or collagen at the indicated concentrations in the presence of Luciferase-Luciferin reagent, and ATP release was measured in a Lumi-aggregometer. (C) The dashed line represents separate analyses. The results are expressed as mean amount of ATP released (nmoles) ± SEM (at least 4 experiments for each condition). #P ≤ .05 between ACC WT and DKI platelets. The data underwent 2-way ANOVA. (D) Representative traces of ATP secretion after 0.5 µg/mL, 1.5 µg/mL, and 2.5 µg/mL collagen stimulation. (E) Washed platelets (30 × 103/µL) were stimulated with 30 mU/mL thrombin or 5 µg/mL collagen for 5 minutes, and serotonin (5-HT) was measured in the supernatant by ELISA kit. The dashed line represents separate analyses. The results are normalized to ACC WT-stimulated platelets and are expressed as means ± SEM (at least 3 experiments for each condition). #P ≤ .05 between ACC WT and DKI platelets. The data underwent 2-way ANOVA. (F) Washed platelets (7.5 × 103/µL) were centrifuged, the pellet was lysed, and serotonin (5-HT) was assayed in the lysate. The results are expressed as means ± SEM (n = 3). (G) Washed platelets were centrifuged, the pellet was lysed, and ADP and ATP content assayed in the lysate by reverse-phase high-performance liquid chromatography. Results are expressed as means ± SEM (n = 3). (H) Washed platelets were stimulated with 100 mU/mL thrombin alone or preincubated or not for 45 minutes with 1 mM aspirin (ASA), and stimulated with 5 µg/mL collagen. TXA2 was measured in the supernatant by ELISA. The dashed line represents separate analyses. The results are expressed as means ± SEM (at least 3 experiments for each condition). #P ≤ .05 between ACC WT and DKI platelets. The data underwent 2-way ANOVA. (I) Washed platelets were preincubated or not for 45 minutes with 1 mM ASA and stimulated with 5 µg/mL collagen. Serotonin (5-HT) was measured in the supernatant by ELISA. The results are expressed as means ± SEM (at least 3 experiments for each condition). #P ≤ .05 between ACC WT and DKI platelets. The data were assessed by 2-way ANOVA. (J) Washed platelets were preincubated with 30 µM ticagrelor or the corresponding vehicle (dimethyl sulfoxide) for 30 minutes and stimulated with 5 µg/mL collagen for 5 minutes. TXA2 was measured in the supernatant by ELISA. The results are expressed as means ± SEM (n = 4). *P ≤ .05 relative to respective untreated platelets, #P ≤ .05 between ACC WT and ACC DKI platelets. The data underwent 2-way ANOVA. See also supplemental Figures 1-5.
ACC DKI platelets display increased dense granule secretion and thromboxane generation on collagen stimulation. (A) Washed platelets were stimulated with thrombin or collagen at the indicated concentrations, and light transmission was measured (Chrono-Log). Aggregation is expressed as the maximal percentage of light transmitted. The dashed line represents separate analyses. The results are expressed as means ± SEM (at least 4 experiments for each condition). (B) αIIbβ3 activation (binding of JON/A) was analyzed by flow cytometry in washed ACC WT and DKI platelets stimulated with thrombin for 8 minutes or collagen for 30 minutes at the indicated concentrations. The dashed line represents separate analyses. The results are expressed as mean fluorescence intensity (MFI) ± SEM (at least 4 experiments for each condition). (C-D) Washed ACC WT and DKI platelets were stimulated with thrombin or collagen at the indicated concentrations in the presence of Luciferase-Luciferin reagent, and ATP release was measured in a Lumi-aggregometer. (C) The dashed line represents separate analyses. The results are expressed as mean amount of ATP released (nmoles) ± SEM (at least 4 experiments for each condition). #P ≤ .05 between ACC WT and DKI platelets. The data underwent 2-way ANOVA. (D) Representative traces of ATP secretion after 0.5 µg/mL, 1.5 µg/mL, and 2.5 µg/mL collagen stimulation. (E) Washed platelets (30 × 103/µL) were stimulated with 30 mU/mL thrombin or 5 µg/mL collagen for 5 minutes, and serotonin (5-HT) was measured in the supernatant by ELISA kit. The dashed line represents separate analyses. The results are normalized to ACC WT-stimulated platelets and are expressed as means ± SEM (at least 3 experiments for each condition). #P ≤ .05 between ACC WT and DKI platelets. The data underwent 2-way ANOVA. (F) Washed platelets (7.5 × 103/µL) were centrifuged, the pellet was lysed, and serotonin (5-HT) was assayed in the lysate. The results are expressed as means ± SEM (n = 3). (G) Washed platelets were centrifuged, the pellet was lysed, and ADP and ATP content assayed in the lysate by reverse-phase high-performance liquid chromatography. Results are expressed as means ± SEM (n = 3). (H) Washed platelets were stimulated with 100 mU/mL thrombin alone or preincubated or not for 45 minutes with 1 mM aspirin (ASA), and stimulated with 5 µg/mL collagen. TXA2 was measured in the supernatant by ELISA. The dashed line represents separate analyses. The results are expressed as means ± SEM (at least 3 experiments for each condition). #P ≤ .05 between ACC WT and DKI platelets. The data underwent 2-way ANOVA. (I) Washed platelets were preincubated or not for 45 minutes with 1 mM ASA and stimulated with 5 µg/mL collagen. Serotonin (5-HT) was measured in the supernatant by ELISA. The results are expressed as means ± SEM (at least 3 experiments for each condition). #P ≤ .05 between ACC WT and DKI platelets. The data were assessed by 2-way ANOVA. (J) Washed platelets were preincubated with 30 µM ticagrelor or the corresponding vehicle (dimethyl sulfoxide) for 30 minutes and stimulated with 5 µg/mL collagen for 5 minutes. TXA2 was measured in the supernatant by ELISA. The results are expressed as means ± SEM (n = 4). *P ≤ .05 relative to respective untreated platelets, #P ≤ .05 between ACC WT and ACC DKI platelets. The data underwent 2-way ANOVA. See also supplemental Figures 1-5.
In addition to αIIbβ3 activation, TXA2 generation and ADP released from dense granules are important players in collagen-induced platelet aggregate formation under flow27,28 and in thrombus formation in vivo.29,30 Interestingly, the lack of AMPK-ACC signaling amplified dense granule release, specifically in response to collagen. Indeed, a significant 30% increase of ATP (Figure 3C-D) and serotonin secretion (Figure 3E) was detected after collagen stimulation in ACC DKI platelets compared with WT, but not in response to thrombin (Figure 3C,E). To further conclude that this increase was related to secretion rather than to augmented packaging of dense granules, we measured total ADP, ATP, and serotonin content in platelet extracts and found no difference between genotypes (Figure 3F-G). Regarding α-granule secretion, P-selectin surface exposure was similar between ACC WT and ACC DKI platelets (supplemental Figure 3A). However, platelet factor 4 (PF4) secretion was potentiated on collagen stimulation in ACC DKI platelets (supplemental Figure 3B), although total PF4 levels in α-granules showed no change (supplemental Figure 3C), indicating that the ACC DKI phenotype seems also associated with increased α-granule secretion, at least those α-granules containing PF4.
Lack of AMPK-ACC signaling potentiated TXA2 generation in response to collagen (Figure 3H), independent of any change in basal cyclooxygenase 1 expression or activity (supplemental Figure 4A-B). To evaluate the connection between ACC DKI-related increased TXA2 generation and dense granule secretion, collagen-stimulated ACC DKI platelets were pretreated with 1 mM aspirin before assessment of serotonin secretion. As expected, cyclooxygenase 1 inhibition by aspirin almost completely abolished collagen-induced TXA2 generation (Figure 3H). In this condition, collagen failed to further enhance serotonin secretion in ACC DKI compared with WT platelets (Figure 3I), demonstrating that the increase in dense granule secretion observed in ACC DKI platelets depends on TXA2 synthesis. The increased TXA2 generation in ACC DKI platelets did not result from the enhanced dense granule release induced by collagen, as ticagrelor, a P2Y12 inhibitor, preserved TXA2 potentiation in ACC DKI platelets (Figure 3J). More interestingly, whole-blood flow perfusion experiments over type I collagen showed that ticagrelor completely abolished the increased thrombus buildup in ACC DKI blood compared with WT, demonstrating the key implication of dense granule secretion in the enhanced thrombus formation (Figure 4A-B).
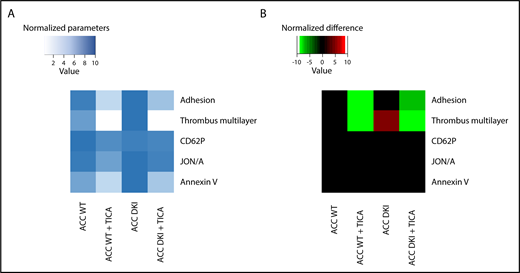
Increased thrombus formation in ACC DKI mice is prevented by P2Y12 inhibition. (A-B) Whole-blood flow perfusion experiments were performed with collagen type I as platelet-adhesive substrate. Where indicated, autocrine effects were blocked by preincubation with 20 µM ticagrelor (TICA) or vehicle (dimethyl sulfoxide) for 10 minutes. Microscopic images were analyzed for indicated parameters, and per parameter normalized on scale from 0 to 10.24 (A) Heat map of normalized parameters. (B) Subtraction heat map of normalized differences compared with WT mice, filtered for changes with P ≤ .05.
Increased thrombus formation in ACC DKI mice is prevented by P2Y12 inhibition. (A-B) Whole-blood flow perfusion experiments were performed with collagen type I as platelet-adhesive substrate. Where indicated, autocrine effects were blocked by preincubation with 20 µM ticagrelor (TICA) or vehicle (dimethyl sulfoxide) for 10 minutes. Microscopic images were analyzed for indicated parameters, and per parameter normalized on scale from 0 to 10.24 (A) Heat map of normalized parameters. (B) Subtraction heat map of normalized differences compared with WT mice, filtered for changes with P ≤ .05.
In addition to thrombin and collagen, ACC WT and ACC DKI platelets were also treated with increasing concentrations of ADP, U46619, rhodocytin, an agonist of the C-type lectin such as receptor-2, or collagen-related peptide, a specific GPVI agonist. For all agonists and concentrations tested, ACC DKI platelets aggregated normally (supplemental Figure 5A-B), and there was no difference in ATP released from dense granules (supplemental Figure 5C). These data reinforce the conclusion that the effect of AMPK-ACC signaling on TXA2 generation and dense granule secretion is specific to platelet response to collagen. Moreover, these results reveal that collagen increases TXA2 generation in ACC DKI platelets, most likely through a mechanism involving the α2β1 integrin, which is compatible with the enhancing role of this integrin in GPVI-dependent thrombus stability via TXA2 production.31
Taken together, these results indicate that the absence of AMPK-ACC signaling in collagen-stimulated platelets elicits increased TXA2 generation and, subsequently, enhanced granule secretion. These events might contribute to exacerbated thrombus growth via a mechanism that is independent of an altered αIIbβ3 activation.
Lack of AMPK-ACC signaling does not affect lipid oxidation, but results in an increased AA-containing PL pool
To test whether persistent activation of ACC in platelets may affect platelet bioenergetics, we measured the thrombin- or collagen-dependent increase in mitochondrial function in ACC WT and ACC DKI platelets. As illustrated in Figure 5A-F, extracellular flux analysis showed that platelet basal oxygen consumption rate (OCR) was similar between ACC WT and DKI mice, and similar to reported studies in the literature.1,20 As expected, thrombin stimulated OCR (Figure 5A-B), which has been shown to be a result of increased mitochondrial lipid oxidation.1,20 The oxidative phosphorylation component was further assessed by injecting oligomycin, a complex V inhibitor, which led to an expected decrease in OCR. ATP-linked respiration was not different in ACC DKI platelets compared with WT platelets. Carbonyl cyanide p-trifluoromethoxyphenylhydrazone, a proton-ionophore, then induced maximal OCR and a corresponding decrease in maximal respiration and reserve capacity on thrombin stimulation, as a result of the mobilization of endogenous metabolic substrates to cope with increased energetic demands. This occurred to a similar extent in platelets from both genotypes (Figure 5A-B). Finally, antimycin A and rotenone, complex III/I inhibitors, totally inhibited mitochondrial-induced OCR. Proton leak was unchanged in all groups (Figure 5A-B). It has not been previously reported that collagen also stimulates mitochondrial function using this approach. As shown in Figure 5C-D, collagen stimulates OCR with similar characteristics to thrombin. Taken together, these data indicate that in platelets, AMPK-ACC signaling is not playing a major role in mitochondrial respiration, both basally and in the presence of thrombin or collagen. The comparable impact of Etomoxir, a carnitine palmitoyltransferase-1 inhibitor, on the ACC WT and ACC DKI platelet mitochondrial bioenergetics further supports the conclusion that platelets from both genotypes similarly rely on fatty acids to produce ATP (Figure 5E-F).
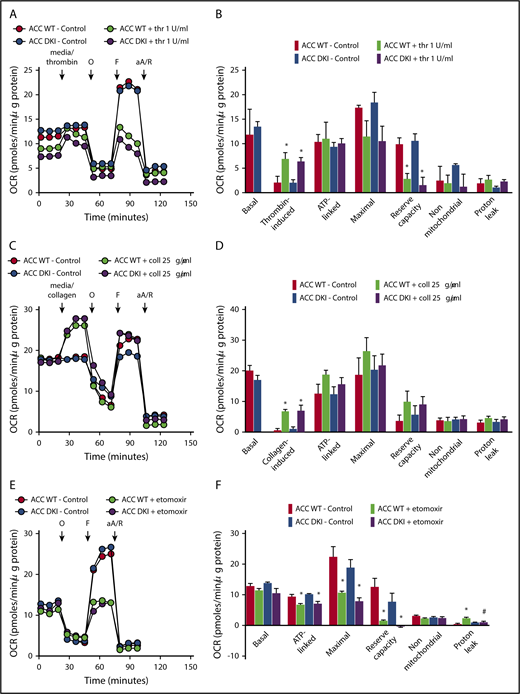
Lack of ACC phosphorylation does not affect oxidative metabolism. (A-F) Oxygen consumption rate (OCR) was measured in washed platelets pretreated or not with Etomoxir (25 μM) (E-F) for 1 hour before bioenergetic measurements. OCR was assessed under basal conditions, after injection of media alone or 1 U/mL thrombin (A,B) or 25 μg/mL collagen (C-D) or followed by treatment with 1 μM oligomycin (O), 0.1 μM carbonyl cyanide p-trifluoromethoxyphenylhydrazone (F), and a mix of 1 μM antimycin A (aA) and 1 μM rotenone (R). (A,C,E) Representative OCR profiles with thrombin (A) and collagen stimulation (C) or Etomoxir treatment (E). (B,D) Mitochondrial function was assessed by calculating basal, thrombin-induced (B) or collagen-induced (D), ATP-linked, maximal, nonmitochondrial, reserve capacity and proton leak OCR. In addition, bioenergetics measurements were assessed in Etomoxir-treated platelets (F). The results are expressed as means ± SEM (at least n = 3). *P ≤ .05 relative to respective untreated platelets, #P ≤ .05 between ACC WT and ACC DKI platelets. The data underwent 2-way ANOVA.
Lack of ACC phosphorylation does not affect oxidative metabolism. (A-F) Oxygen consumption rate (OCR) was measured in washed platelets pretreated or not with Etomoxir (25 μM) (E-F) for 1 hour before bioenergetic measurements. OCR was assessed under basal conditions, after injection of media alone or 1 U/mL thrombin (A,B) or 25 μg/mL collagen (C-D) or followed by treatment with 1 μM oligomycin (O), 0.1 μM carbonyl cyanide p-trifluoromethoxyphenylhydrazone (F), and a mix of 1 μM antimycin A (aA) and 1 μM rotenone (R). (A,C,E) Representative OCR profiles with thrombin (A) and collagen stimulation (C) or Etomoxir treatment (E). (B,D) Mitochondrial function was assessed by calculating basal, thrombin-induced (B) or collagen-induced (D), ATP-linked, maximal, nonmitochondrial, reserve capacity and proton leak OCR. In addition, bioenergetics measurements were assessed in Etomoxir-treated platelets (F). The results are expressed as means ± SEM (at least n = 3). *P ≤ .05 relative to respective untreated platelets, #P ≤ .05 between ACC WT and ACC DKI platelets. The data underwent 2-way ANOVA.
To investigate whether the gain-of-function of ACC DKI platelets relies on a modified lipid content, we undertook a quantitative lipidomic analysis of resting and stimulated platelets. Isolated ACC DKI and ACC WT platelets were either left unstimulated or activated with 25 µg/mL collagen, 0.3 U/mL thrombin, or a mix of both. The lipids classes analyzed included different PL (PE, PEP, plasmenyl phosphatidylethanolamine, phosphatidylcholine [PC], lysophosphatidylethanolamine, lysophosphatidylcholine), sphingomyelin, cholesteryl esters, free fatty acids, DAG, and triglycerides.32
In agreement with previous data,33 class enrichment analysis indicated that collagen significantly upregulated lipid species among free fatty acids (31%), lysophosphatidylcholine (57%), lysophosphatidylethanolamine (38%), and DAG (21%) lipid classes, in both ACC WT and ACC DKI platelets (Figure 6A; supplemental Table 2). Enrichment of free fatty acids and DAG was also observed in response to thrombin alone (supplemental Figure 6A) and to a combination of thrombin and collagen (supplemental Figure 6B). More important, loss of AMPK-ACC signaling resulted in enrichment of upregulated lipid species among PEP lipid classes (28%), independent of the basal or simulated condition (Figure 6B; supplemental Table 3). Indeed, an increase in lipid species among PEP was strongly associated with lack of ACC phosphorylation (odds ratio, 48 [CI, 14-217]; Figure 6B). ACC DKI also displayed an increment of downregulated lipid species among cholesteryl esters (38%) and PC (14%; Figure 6B). Given that ACC DKI platelets show increased TXA2 generation, we specifically focused our data analysis on AA-containing PEP and PC, the 2 PL classes particularly containing differentially regulated lipids. Relevant PEP species with side-chains 16:0/20:4 and 18:1/20:4 were significantly increased in ACC DKI platelets (Figure 6C). We showed that they, respectively, contributed 18.9% and 7.3% to the total reservoir of AA-containing PL (Figure 6D). In contrast, PC species with side-chains 18:2/20:4 and 14:0/20:4 were decreased (Figure 6C) and marginally accounted for 0.8% and 0.4%, respectively, of all AA-containing PL (Figure 6D). Taken together, upregulated AA-containing PEP in ACC DKI platelets clearly supports the enhanced TXA2 formation observed in this condition. Of note, the AMPK-ACC signaling did not influence platelet palmitate uptake (supplemental Figure 7A). Moreover, collagen-induced TXA2 production was not modulated by extracellular fatty acid availability in ACC WT or ACC DKI platelets (supplemental Figure 7B).
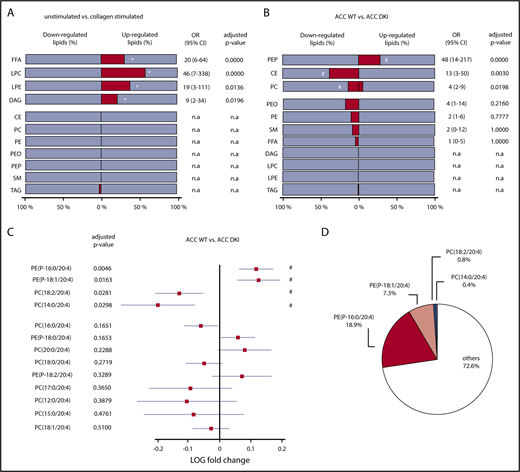
Lack of AMPK-ACC signaling results in an increased AA-containing PL pool. (A,B) The percentage (dark gray bar) of down- or upregulated lipid species relative to the whole lipid class was calculated in collagen-stimulated platelets relative to unstimulated platelets (A) or ACC DKI relative to ACC WT platelets (B). The different lipid classes are arranged from top down in order of increasing adjusted P values. (A) *Adjusted P ≤ .05 between collagen-stimulated platelets and unstimulated platelets (n = 3). (B) #P ≤ .05 between ACC DKI and WT platelets (n = 3). Multivariate regression analysis was performed. (C) Comparison of AA-containing PEP and PC between ACC DKI and WT platelets. The data are presented as log fold change (squares) relative to ACC WT platelets and with 95% confidence interval (CI) (horizontal lines). Statistical significance was based on false discovery rate P values (adjusted P values). The different lipid species are arranged from top down in order of increasing adjusted P values. Multivariate regression analysis was performed. #P ≤ .05 between ACC WT and ACC DKI platelets. (D) Proportions of PE(P-16:0/20:4), PE(P-18:1/20:4), PC(18:2/20:4), and PC(14:0/20:4) relative to all AA-containing PL were evaluated in ACC WT platelets. See also supplemental Figure 6 and 7 and supplemental Table 2 and 3. OR, odds ratio; n.a., not applicable (if no change or if change is OR < 1).
Lack of AMPK-ACC signaling results in an increased AA-containing PL pool. (A,B) The percentage (dark gray bar) of down- or upregulated lipid species relative to the whole lipid class was calculated in collagen-stimulated platelets relative to unstimulated platelets (A) or ACC DKI relative to ACC WT platelets (B). The different lipid classes are arranged from top down in order of increasing adjusted P values. (A) *Adjusted P ≤ .05 between collagen-stimulated platelets and unstimulated platelets (n = 3). (B) #P ≤ .05 between ACC DKI and WT platelets (n = 3). Multivariate regression analysis was performed. (C) Comparison of AA-containing PEP and PC between ACC DKI and WT platelets. The data are presented as log fold change (squares) relative to ACC WT platelets and with 95% confidence interval (CI) (horizontal lines). Statistical significance was based on false discovery rate P values (adjusted P values). The different lipid species are arranged from top down in order of increasing adjusted P values. Multivariate regression analysis was performed. #P ≤ .05 between ACC WT and ACC DKI platelets. (D) Proportions of PE(P-16:0/20:4), PE(P-18:1/20:4), PC(18:2/20:4), and PC(14:0/20:4) relative to all AA-containing PL were evaluated in ACC WT platelets. See also supplemental Figure 6 and 7 and supplemental Table 2 and 3. OR, odds ratio; n.a., not applicable (if no change or if change is OR < 1).
Discussion
Lipids play fundamental roles in platelets, but little is known about the effect of endogenous lipid metabolism on platelet functions and subsequent thrombosis. To assess the importance of endogenous lipid synthesis on platelet function, we characterized a mouse model carrying a genetic mutation that prevents the AMPK-induced phosphorylation of ACC, the first rate-limiting enzyme of lipid synthesis.2 We have shown that ACC1 is the predominant isoform in murine and human platelets. These data are consistent with transcriptomic and proteomic analyses that demonstrate ACC1 but not ACC2 transcript and protein expression in platelets.34,35 In accordance with the primary role of ACC1 in lipogenesis, ACC DKI platelets display modified PL content rather than altered lipid oxidation. Indeed, no difference in oxygen consumption, which relies on endogenous lipid oxidation,20 is observed between ACC WT and ACC DKI platelets under both basal conditions and on thrombin or collagen treatment. Consistent with our results, liver-specific inactivation of ACC1 resulted in reduced de novo fatty acid synthesis without alteration of fatty acid oxidation.36
In the first series of experiments, we demonstrated that bleeding time is shorter in ACC DKI mice and thrombosis is increased in vivo and ex vivo under flow on collagen. Second, platelet functions were analyzed and the results indicate that ACC DKI platelets exhibit enhanced dense granule secretion in response to collagen, attributed to exacerbated TXA2 generation. Finally, untargeted lipidomics revealed that lack of AMPK-ACC signaling was associated with an increase in some AA-containing PEP, leading to TXA2 enrichment. Our findings highlight the critical role of platelet endogenous lipid synthesis in thrombosis and hemostasis, a concept that is supported by a recent study showing that a genetic deletion of acid sphingomyelinase, which converts sphingomyelin to ceramide, leads to an alteration of platelet lipidome, which in turn causes dysregulated granule secretion and in vitro thrombus formation.37
Thrombosis induced by FeCl3 in the carotid artery is a widely used model that can provide valuable information about the effect of genetic modifications on platelet functions. However, it has a number of limitations regarding the underlying mechanisms that induce thrombus formation, notably about the relative roles of tissue factor and thrombin.38-41 Recently, several studies reported that plasma proteins and blood cells, including platelets, aggregate because their negatively charged proteins bind to positively charged iron species.42,43 Altogether, these results indicate that FeCl3-induced thrombosis relies on complex multifaceted, incompletely elucidated, mechanisms.
Collectively, the data presented here demonstrate that AMPK-induced ACC1 phosphorylation in platelets does not control lipid oxidation but, rather, regulates cellular lipid content. Intriguingly, we expect that an increase in AA-containing PL, as observed in ACC DKI platelets, would provide more substrates for mitochondrial respiration in response to thrombin.1 However, we could not find any modifications of platelet mitochondrial oxygen consumption, indicating some degree of compartmentalization and suggesting that increased PEP16:0/20:4 and 18:1/20:4 in ACC DKI platelets do not significantly contribute to overall platelet bioenergetics.
The view that TXA2 and dense granules are of central importance in triggering thrombus formation but not platelet activation on collagen surfaces has been advanced in previous studies.27,28 In addition, the relationship between TXA2 and dense granule secretion has been clearly established.15,44,45 Our work not only confirms this link in ACC DKI platelets but also underlines that excessive thrombus formation is not systematically associated with an alteration of αIIbβ3-dependent platelet aggregation. This disconnection has been recently reported37 and supports the notion that additional cell surface molecules participate in platelet/platelet interactions and support thrombus growth.46,47 Therefore, based on the core-shell thrombosis model,48,49 we postulate that lack of AMPK-ACC signaling might potentiate platelet recruitment to the shell structure through increased TXA2 and ADP secretion, augmenting thrombosis through mechanisms independent of αIIbβ3. In addition, increased PF4 secretion might possibly contribute to a rise in thrombus growth in ACC DKI mice.
Interestingly, the effect of AMPK-ACC signaling on TXA2 generation and platelet dense granule secretion is collagen-specific. This is in agreement with previous data showing that different mechanisms of AA-containing PL breakdown are operating in response to collagen or thrombin. Indeed, although AA release by thrombin is dependent on cPLA2,1,50 collagen-induced AA generation involves cPLA2, the low-molecular-weight secreted PLA2 (sPLA2) and the Ca2+-independent cytosolic PLA2 (iPLA2).51 In line with these observations, thrombin and collagen differentially degrade AA-containing PL, depending on the nature of the PL.37,52 For instance, thrombin is more potent at cleaving AA-containing PI and phosphatidylserine than collagen, whereas the latter is 2 times more efficient toward AA-containing PEP.52 These data reinforce our observations and help us to understand the specific response or responses of ACC DKI platelets to collagen.
Importantly, ACC phosphorylation could be detected, even in the absence of any agonist stimulation, in platelets from both healthy mice and volunteers.7 This has been confirmed by comparing ACC phosphorylation between ACC WT and ACC DKI platelets under basal conditions. A sustained change in basal ACC phosphorylation was sufficient to induce a decrease in PL, suggesting that ACC phosphorylation already affects lipid composition in resting platelets before activation.
In conclusion, our work provides new insights into the contribution of endogenous lipid synthesis to platelet functions. It reveals that sustained modulation of ACC phosphorylation in platelets can modify specific AA-containing PL content and influence platelet reactivity to collagen. This finding might have far-reaching clinical relevance in the pathological context of diseases such as atherosclerosis. Indeed, this is now being tested in a clinical trial demonstrating that basal ACC phosphorylation dramatically increases in platelets of patients with coronary artery disease (ACCTHEROMA Clinical Trial identifier: NCT03034148). How and whether such a pathway is involved in regulating platelet function in this context remain to be determined.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Pierre Sonveaux (Pole of Pharmacology, Institut de Recherche Expérimentale et Clinique, Université catholique de Louvain, Brussels, Belgium) for his help with extracellular flux analysis of mitochondrial function.
This work was supported by grants from Fonds National de la Recherche Scientifique et Médicale (FNRS, Brussels, Belgium), Action de Recherche Concertée de la Communauté Wallonie-Bruxelles, Brussels, Belgium (ARC 13/18-051, ARC 16-21), and Fondation Louvain, Brussels, Belgium, and with unrestricted grants from Bayer and Astra Zeneca. S.L. and S.K. were supported by FNRS. S.L. and M.-B.O. were supported by Bourse Salus Sanguinis (Université catholique de Louvain). M.O. and O.W. have a Fonds pour la formation à la Recherche dans l'Industrie et l'Agriculture (FRIA) fellowship (FNRS). B.E.K. is supported by the National Health and Medical Research Council and the Victorian Government Operational Infrastructure Support Scheme. S.B. and J.H. are supported by the Interreg Euregio Meuse-Rhine program Polyvalve. S.H. is research associate, and L.B. and C.O. are senior research associates at FNRS. C.B. was a clinical master specialist at FNRS.
Authorship
Contribution: S.L., M.-B.O., L.B., C.O., C.B., and S.H. provided conceptualization; V.M.D.-U. provided methodology; M.G., J.A., and B.G. provided formal analysis; S.L., S.K., M.O., A.G., M.-B.O., A.H., O.W., S.B., F.S., M.G., and J.H. performed the investigation; G.R.S., B.E.K., and C.O. provided resources; S.L., V.M.D.-U., C.B., and S.H., wrote the original draft and performed a review; and C.B. and S.H. performed supervision.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Sandrine Horman, Pôle de Recherche Cardiovasculaire, Institut de Recherche Expérimentale et Clinique, Université catholique de Louvain, 55, Avenue Hippocrate, 1200 Brussels, Belgium; e-mail: sandrine.horman@uclouvain.be.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal