

TO THE EDITOR:
The pathophysiologic mechanisms, genetic predictors, and long-term consequences of acute kidney injury (AKI) on kidney function in sickle cell anemia (SCA) are not clear.1-5 In the general population, AKI is associated with the subsequent development and progression of chronic kidney disease (CKD).6,7 Animal models have demonstrated that AKI results in capillary loss and tissue hypoxia, dysregulated apoptosis, and sustained proinflammatory and profibrotic signaling.7 We investigated clinical and laboratory predictors for AKI in a cohort of hospitalized adults with SCA at the University of Illinois at Chicago. We also examined whether genetic variants implicated in sickle cell nephropathy8-10 predicted AKI risk and whether AKI is associated with a more rapid decline in kidney function on longitudinal follow-up.
We analyzed 137 SCA (HbSS/Sβ0-thalassemia) patients enrolled between August of 2010 and June of 2012 into a prospective registry and followed until December of 2017. These patients were selected from a cohort of 267 SCA patients based on genotyping availability and hospitalization during the observation period. Patients included in this analysis were similar to those not included with respect to age, sex, hydroxyurea use, estimated glomerular filtration rate (eGFR), and albuminuria at enrolment (P ≥ .2). The protocol was approved by the Institutional Review Board before initiating the study, and all subjects provided written informed consent.
Clinical and laboratory data were collected at the time of hospitalization for the first AKI event or the first vaso-occlusive crisis in those without an AKI event. AKI was defined according to the Kidney Disease Improving Global Guidelines as a rise in serum creatinine ≥0.3 mg/dL within 48 hours or a >50% rise in serum creatinine within 7 days.11 AKI severity was staged according to the Kidney Disease Improving Global Guidelines as follows: stage 1 = serum creatinine rise 1.5-1.9 times baseline, stage 2 = serum creatinine rise 2.0-2.9 times baseline, and stage 3 = serum creatinine rise ≥3 times baseline, serum creatinine ≥4.0 mg/dL, or requiring hemodialysis. The CKD-Epidemiology Collaboration formula was used to calculate eGFR.12 CKD progression and hemoglobinuria were defined as previously described.13 A single systolic blood pressure measured during emergency room triaging and prior to initiating therapy was used for the analyses. Genotyping for the APOL1 G1/G2 risk variants, HMOX1 rs743811 and GT tandem repeats, and α-thalassemia was conducted as previously described.10,14
Linear and categorical variables were compared by AKI status using the Mann-Whitney or χ2 test, respectively. Applying the Bonferroni correction, P < .004 was considered statistically significant. Variables with P ≤ .1 were entered into a logistic regression model, and a step-wise approach was used to determine the final AKI risk model. A generalized estimating equation was applied to compare eGFR slope pre-AKI with post-AKI with the fully adjusted model including age, sex, and hydroxyurea. Rates of CKD progression were compared by AKI status and AKI stage using the log-rank method and Cox proportional hazards ratio, adjusted for age, sex, hydroxyurea, and baseline eGFR.
With a median follow-up of 68 months (interquartile range [IQR], 51-74 months), 135 AKI events were observed during 2691 (5%) hospitalizations. The clinical settings for an AKI event are provided in supplemental Table 1 (available on the Blood Web site), and the severity of the 135 AKI events are provided in Figure 1A. Three patients with stage 3 AKI required hemodialysis during the AKI event. The eGFR did not return to baseline values prior to the AKI event in 21 of the 135 (16%) AKI events (supplemental Figure 1).
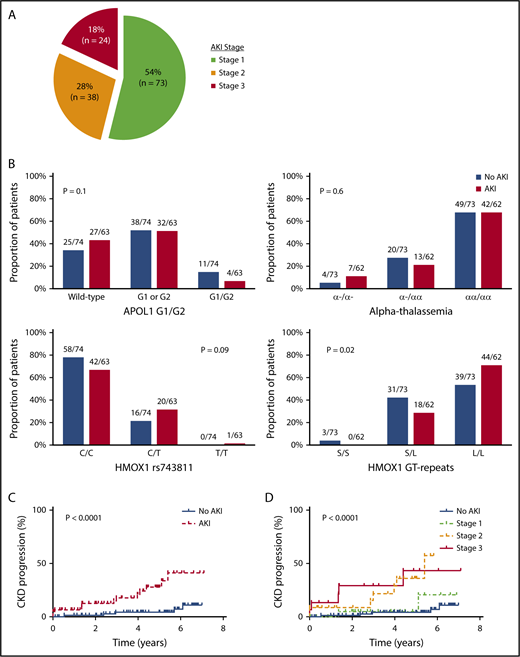
AKI in SCA. (A) Severity of AKI events among patients with SCA. AKI severity was stage 1 (serum creatinine rise 1.5-1.9 times baseline) in 73 of 135 (54%) AKI events, stage 2 (serum creatinine rise 2.0-2.9 times baseline) in 38 of 135 (28%) AKI events, and stage 3 (serum creatinine rise ≥3 times baseline, serum creatinine ≥4.0 mg/dL, or requiring hemodialysis) in 24 of 135 (18%) AKI events observed in patients with SCA during the follow-up period. (B) AKI risk by candidate gene variants. No association for AKI risk with APOL1 G1 and G2 risk variants or coinheritance of α-thalassemia was observed, whereas trends were observed for an association with HMOX1 rs743811 T risk allele and long GT tandem repeats in the promoter region of HMOX1. (C) CKD progression by AKI status. SCA patients with an AKI event (14/63, 22%) had more rapid CKD progression (defined as a 50% reduction in eGFR or requiring renal replacement therapy) compared with those without an AKI event (5/74, 7%). (D) CKD progression by AKI severity. Severity of AKI events was associated with more rapid progression of CKD in patients with SCA (stage 3: 5/15, 33%; stage 2: 7/22, 32%; stage 1: 2/26, 8%; no AKI: 5/74, 7%).
AKI in SCA. (A) Severity of AKI events among patients with SCA. AKI severity was stage 1 (serum creatinine rise 1.5-1.9 times baseline) in 73 of 135 (54%) AKI events, stage 2 (serum creatinine rise 2.0-2.9 times baseline) in 38 of 135 (28%) AKI events, and stage 3 (serum creatinine rise ≥3 times baseline, serum creatinine ≥4.0 mg/dL, or requiring hemodialysis) in 24 of 135 (18%) AKI events observed in patients with SCA during the follow-up period. (B) AKI risk by candidate gene variants. No association for AKI risk with APOL1 G1 and G2 risk variants or coinheritance of α-thalassemia was observed, whereas trends were observed for an association with HMOX1 rs743811 T risk allele and long GT tandem repeats in the promoter region of HMOX1. (C) CKD progression by AKI status. SCA patients with an AKI event (14/63, 22%) had more rapid CKD progression (defined as a 50% reduction in eGFR or requiring renal replacement therapy) compared with those without an AKI event (5/74, 7%). (D) CKD progression by AKI severity. Severity of AKI events was associated with more rapid progression of CKD in patients with SCA (stage 3: 5/15, 33%; stage 2: 7/22, 32%; stage 1: 2/26, 8%; no AKI: 5/74, 7%).
Sixty-three of 137 (46%) SCA patients had an AKI event during the follow-up period. SCA patients with an AKI event were older, had a higher white blood cell (WBC) count, a higher serum creatinine concentration, a greater prevalence of hemoglobinuria at the time of hospitalization, and were more frequently treated with vancomycin during the hospitalization compared with hospitalized SCA patients without an AKI event (P ≤ .001) (Table 1). HMOX1 long GT tandem repeats, but not APOL1 G1/G2 risk variants or α-thalassemia, were associated with increased occurrence of AKI (Figure 1B). On logistic-regression analysis, older age (10-year increase: odds ratio [OR], 3.6; 95% confidence interval [CI], 2.0-6.2; P < .0001), lower systolic blood pressure (10-mmHg decrease: OR, 1.8; 95% CI, 1.3-2.6; P = .0006), higher WBC count at the time of hospitalization (OR, 1.2; 95% CI, 1.1-1.3; P = .005), long HMOX1 GT tandem repeats (OR, 4.1; 95% CI, 1.5-11.4; P = .007), vancomycin use (OR, 4.5; 95% CI, 1.2-16.7; P = .02), and the number of hospitalizations (10-hospitalization increase: OR, 1.3; 95% CI, 1.02-1.7; P = .03) were independently associated with the occurrence of AKI.
Characteristics associated with AKI in hospitalized patients with SCA
| Variables | N | No AKI | N | AKI | P |
|---|---|---|---|---|---|
| At the time of hospitalization | |||||
| Age, median (IQR), y | 74 | 30 (24-37) | 63 | 38 (28-48) | <.0001 |
| Total number of hospitalizations, median (IQR) | 74 | 9 (3-25) | 63 | 17 (6-29) | .08 |
| Diabetes mellitus history, n (%) | 74 | 2 (3) | 63 | 2 (3) | .9 |
| Hypertension history, n (%) | 74 | 8 (11) | 63 | 10 (16) | .4 |
| Hydroxyurea use, n (%) | 74 | 34 (46) | 63 | 34 (54) | .5 |
| ACE inhibitor or ARB therapy, n (%) | 74 | 12 (16) | 63 | 12 (19) | .7 |
| Outpatient NSAID use, n (%) | 74 | 23 (31) | 63 | 17 (27) | .6 |
| Chronic transfusion therapy, n (%) | 74 | 9 (12) | 63 | 12 (19) | .3 |
| Systolic blood pressure, median (IQR), mm Hg | 74 | 122 (113-140) | 63 | 116 (106-127) | .008 |
| WBC count, median (IQR), ×103/µL | 74 | 11.1 (9.0-14.6) | 63 | 13.7 (11.0-17.2) | .001 |
| Hemoglobin, median (IQR), g/dL | 74 | 8.5 (7.3-9.6) | 63 | 7.9 (7.1-8.6) | .02 |
| Indirect bilirubin, median (IQR), mg/dL | 55 | 1.9 (1.3-4.0) | 53 | 2.5 (1.5-4.0) | .4 |
| Hemoglobinuria, n (%) | 44 | 9 (20) | 40 | 23 (58) | .0005 |
| Serum creatinine, median (IQR), mg/dL | 74 | 0.7 (0.5-0.8) | 63 | 1.2 (0.8-1.4) | <.0001 |
| Urine albumin, median (IQR), mg/g creatinine | 69 | 38 (12-159) | 58 | 91 (23-317) | .05 |
| Variables | N | No AKI | N | AKI | P |
|---|---|---|---|---|---|
| At the time of hospitalization | |||||
| Age, median (IQR), y | 74 | 30 (24-37) | 63 | 38 (28-48) | <.0001 |
| Total number of hospitalizations, median (IQR) | 74 | 9 (3-25) | 63 | 17 (6-29) | .08 |
| Diabetes mellitus history, n (%) | 74 | 2 (3) | 63 | 2 (3) | .9 |
| Hypertension history, n (%) | 74 | 8 (11) | 63 | 10 (16) | .4 |
| Hydroxyurea use, n (%) | 74 | 34 (46) | 63 | 34 (54) | .5 |
| ACE inhibitor or ARB therapy, n (%) | 74 | 12 (16) | 63 | 12 (19) | .7 |
| Outpatient NSAID use, n (%) | 74 | 23 (31) | 63 | 17 (27) | .6 |
| Chronic transfusion therapy, n (%) | 74 | 9 (12) | 63 | 12 (19) | .3 |
| Systolic blood pressure, median (IQR), mm Hg | 74 | 122 (113-140) | 63 | 116 (106-127) | .008 |
| WBC count, median (IQR), ×103/µL | 74 | 11.1 (9.0-14.6) | 63 | 13.7 (11.0-17.2) | .001 |
| Hemoglobin, median (IQR), g/dL | 74 | 8.5 (7.3-9.6) | 63 | 7.9 (7.1-8.6) | .02 |
| Indirect bilirubin, median (IQR), mg/dL | 55 | 1.9 (1.3-4.0) | 53 | 2.5 (1.5-4.0) | .4 |
| Hemoglobinuria, n (%) | 44 | 9 (20) | 40 | 23 (58) | .0005 |
| Serum creatinine, median (IQR), mg/dL | 74 | 0.7 (0.5-0.8) | 63 | 1.2 (0.8-1.4) | <.0001 |
| Urine albumin, median (IQR), mg/g creatinine | 69 | 38 (12-159) | 58 | 91 (23-317) | .05 |
| During hospitalization | |||||
|---|---|---|---|---|---|
| NSAID use, n (%) | 74 | 27 (36) | 63 | 13 (21) | .04 |
| Intravenous contrast use, n (%) | 74 | 9 (12) | 63 | 6 (10) | .6 |
| Vancomycin use, n (%) | 74 | 5 (7) | 63 | 17 (27) | .001 |
| β-Lactam antibiotic use, n (%) | 74 | 28 (38) | 63 | 24 (38) | .9 |
| Sepsis, n (%) | 74 | 1 (1) | 63 | 2 (3) | .5 |
| During hospitalization | |||||
|---|---|---|---|---|---|
| NSAID use, n (%) | 74 | 27 (36) | 63 | 13 (21) | .04 |
| Intravenous contrast use, n (%) | 74 | 9 (12) | 63 | 6 (10) | .6 |
| Vancomycin use, n (%) | 74 | 5 (7) | 63 | 17 (27) | .001 |
| β-Lactam antibiotic use, n (%) | 74 | 28 (38) | 63 | 24 (38) | .9 |
| Sepsis, n (%) | 74 | 1 (1) | 63 | 2 (3) | .5 |
ACE, angiotensin-converting enzyme; ARB, angiotensin receptor blocker; NSAID, nonsteroidal anti-inflammatory drug.
On longitudinal follow-up, SCA patients with an AKI event had a more rapid decline in eGFR after the AKI event compared with pre-AKI (β = −4.9 ± 1.6 mL/min/1.72 m2 per year; P = .002). CKD progression was observed in 19 of 137 (14%) SCA patients; 15 (79%) were due to a 50% decline in eGFR, and 4 (21%) were due to the requirement of hemodialysis. CKD progression occurred in 14 of 63 (22%) patients with an AKI event and in 5 of 74 (7%) patients without an AKI event (P < .0001, log-rank test) (Figure 1C). Developing an AKI was independently associated with an increased risk for CKD progression (hazard ratio [HR], 4.6; 95% CI, 1.6-13.4; P = .005). More severe episodes of AKI were associated with increased proportions of patients with CKD progression (AKI stage 3: 5/15, 33%; AKI stage 2: 7/22, 32%; AKI stage 1: 2/26, 8%) (P < .0001, log-rank test) (Figure 1D). AKI stage was an independent risk factor for CKD progression after adjustment for age, sex, hydroxyurea, and baseline eGFR (HR, 2.3; 95% CI: 1.5-3.6; P = .0002).
Older age was a risk factor for AKI in this SCA cohort, consistent with the recognized age-related physiological changes that lead to a reduction in renal reserve and increased susceptibility to AKI in other conditions.15,16 Higher WBC counts, lower systolic blood pressure, vancomycin use, and frequent hospitalizations were also risk factors for AKI, consistent with some observations in other sickle cell disease cohorts.1-4 The increased use of vancomycin in SCA patients that developed AKI may reflect the direct tubular toxicity of vancomycin17 or the increased severity of illness during that hospitalization. Nonsteroidal anti-inflammatory drug use was less common during hospitalizations with an AKI event and likely reflects the avoidance of these drugs in the setting of an elevated serum creatinine.
New in our study is the observation that long HMOX1 GT tandem repeats were independently associated with the occurrence of AKI. HMOX1 is the inducible rate-limiting enzyme that catabolizes the pro-oxidant heme. Long GT tandem repeats correlate with reduced HMOX1 messenger RNA expression and HMOX1 activity.18 Taken together with the association of HMOX1 GT tandem repeats with AKI, the higher prevalence of hemoglobinuria in patients who developed an AKI suggests that impaired cell-free hemoglobin processing may be a pathway for AKI in SCA.
Our observation that AKI and AKI severity are independent risk factors for CKD progression in SCA patients is consistent with the clinical importance of AKI and AKI severity in CKD progression that has been demonstrated in patients undergoing cardiac procedures and in the general population.6,7,19-21
Our study is limited by it being the experience of a single center. Larger cohorts may be able to identify other genetic risk variants for AKI in SCA. AKI events prior to enrolment or that occurred outside of our institution may have been missed. Monitoring strict urine output for the additional definition of AKI (urine output <0.5 mL/kg per hour for 6 hours) and adherence to hydroxyurea should be addressed in subsequent studies. Future studies inducing HMOX1 as a renoprotective measure and evaluating AKI in the pathophysiology of sickle cell nephropathy are warranted.
The online version of this article contains a data supplement.
Acknowledgments
The project described was supported by the National Institutes of Health, National Heart, Lung, and Blood Institute through grants K23HL125984 (S.L.S.), R01HL111656 (R.F.M.), R01HL127342 (R.F.M.), R01HL078536 (R.E.M.), and National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases K24DK092290 (J.P.L.) and received statistical support from the National Center for Advancing Translational Sciences National Institutes of Health through grant UL1TR002003.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Authorship
Contribution: S.L.S. designed and performed research, analyzed the data, and wrote the manuscript; M.V., A.R., R.R., B.N.S., X.Z., J.H., M.G., S.J., R.E.M., and R.F.M. performed research and contributed to the data analysis; and J.P.L. and V.R.G. designed the research, analyzed the data, and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Santosh L. Saraf, Division of Hematology-Oncology, Department of Internal Medicine, University of Illinois at Chicago, 820 South Wood St, Suite 172, Chicago, IL 60612; e-mail: ssaraf@uic.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal