

TO THE EDITOR:
The “unclassifiable” myelodysplastic/myeloproliferative neoplasms (MDS/MPN-U) remain the most poorly characterized among the various subtypes of MDS/MPN.1 Retrospective studies have reported the median survival of patients with MDS/MPN-U to be 21 months from diagnosis2 and 12 months from referral to a tertiary care institution.3 We sought to characterize the mutational landscape of MDS/MPN-U through targeted gene sequencing.
Adults with a diagnosis of MDS/MPN-U confirmed at 1 of 4 academic medical centers (MD Anderson Cancer Center [MDACC], Cleveland Clinic, Moffitt Cancer Center, and Vanderbilt University) who had targeted next-generation sequencing (NGS) performed on their bone marrow (BM) aspirates or peripheral blood (PB) using each institution’s multigene panel form the subjects of this study.
At each site, the diagnosis of MDS/MPN-U was confirmed per the 2008 World Health Organization (WHO) criteria (supplemental Table 1, available on the Blood Web site).4 Cases fulfilling the diagnostic criteria for MDS/MPN with ringed sideroblasts and thrombocytosis,5 atypical chronic myeloid leukemia (aCML),2 or chronic neutrophilic leukemia6 were excluded. An extensive workup was performed to exclude nonclonal causes. Targeted NGS was performed on BM aspirates (or PB samples, for some patients at the Cleveland Clinic and most at Moffitt) as previously described.7,8 Supplemental Table 2 lists the gene panels used at each institution. Twenty genes were in common (sequenced in all patients): ASXL1, CBL, DNMT3A, ETV6, EZH2, IDH1, IDH2, JAK2, KIT, NPM1, NRAS, PHF6, RUNX1, SETBP1, SF3B1, SRSF2, TET2, TP53, U2AF1, and ZRSR2.
Data were collected at each institution under an institutional review board–approved retrospective chart review protocol. Overall survival (OS) was analyzed using the Kaplan-Meier method and compared by the log-rank test.
One hundred two patients (48 seen at MDACC, 29 at the Cleveland Clinic, 16 at Moffitt, and 9 at Vanderbilt) had targeted NGS performed on their BM aspirates or PB, generally upon first presentation to the referral center. Baseline characteristics of the patients (as of the sample date) appear in Table 1.
Baseline characteristics of the patients
| Characteristics | No. (%) or median (range) |
|---|---|
| Clinical parameters | |
| Sex | |
| Female | 24 (24) |
| Male | 78 (76) |
| Age, y | 70 (21-89) |
| Leukocyte count, ×109/L | 13.3 (1-179) |
| Absolute neutrophil count, ×109/L | 7.48 (0.39-152.4) |
| Hemoglobin level, g/dL | 9.3 (3.1-15) |
| Platelet count, ×109/L | 120 (6-1168) |
| BM blast percentage, % | 2 (0-17) |
| Time from diagnosis, mo | 3.1 (0-149) |
| Site | |
| MDACC | 48 (47) |
| Cleveland Clinic | 29 (28) |
| Moffitt | 16 (16) |
| Vanderbilt | 9 (9) |
| Cytogenetics: IPSS-R category24 | |
| Very good | 1 (1) |
| Good | 57 (56) |
| Intermediate | 28 (27) |
| Poor | 8 (8) |
| Very poor | 4 (4) |
| Insufficient metaphases/not done | 4 (4) |
| IPSS-R10 : risk category | |
| Very low | 13 (13) |
| Low | 47 (46) |
| Intermediate | 19 (19) |
| High | 15 (15) |
| Very high | 5 (5) |
| Not available | 3 (3) |
| DIPSS11 : risk category | |
| Low | 7 (7) |
| Intermediate-1 | 34 (33) |
| Intermediate-2 | 39 (38) |
| High | 22 (22) |
| Characteristics | No. (%) or median (range) |
|---|---|
| Clinical parameters | |
| Sex | |
| Female | 24 (24) |
| Male | 78 (76) |
| Age, y | 70 (21-89) |
| Leukocyte count, ×109/L | 13.3 (1-179) |
| Absolute neutrophil count, ×109/L | 7.48 (0.39-152.4) |
| Hemoglobin level, g/dL | 9.3 (3.1-15) |
| Platelet count, ×109/L | 120 (6-1168) |
| BM blast percentage, % | 2 (0-17) |
| Time from diagnosis, mo | 3.1 (0-149) |
| Site | |
| MDACC | 48 (47) |
| Cleveland Clinic | 29 (28) |
| Moffitt | 16 (16) |
| Vanderbilt | 9 (9) |
| Cytogenetics: IPSS-R category24 | |
| Very good | 1 (1) |
| Good | 57 (56) |
| Intermediate | 28 (27) |
| Poor | 8 (8) |
| Very poor | 4 (4) |
| Insufficient metaphases/not done | 4 (4) |
| IPSS-R10 : risk category | |
| Very low | 13 (13) |
| Low | 47 (46) |
| Intermediate | 19 (19) |
| High | 15 (15) |
| Very high | 5 (5) |
| Not available | 3 (3) |
| DIPSS11 : risk category | |
| Low | 7 (7) |
| Intermediate-1 | 34 (33) |
| Intermediate-2 | 39 (38) |
| High | 22 (22) |
In 28 cases, the diagnosis of MDS/MPN-U was first made at 1 of the 4 tertiary care institutions.
Mutational frequencies for the 20 genes evaluated in all patients are shown in Figure 1A. The frequencies of mutations in 10 other genes often encountered in myeloid malignancies (CALR, MPL, BRAF, CSF3R, FLT3, PTPN11, MLL, STAG2, CEBPA, and KRAS) are also depicted. Figure 1B additionally illustrates the frequencies of cooccurrence of mutations (restricted to the 20 genes sequenced in all patients) within the same patient. Most mutations occurred at known “hotspots.” The highest variant allele frequencies were found for mutant ASXL1 and TET2 (21% each), followed by JAK2 (19%), SRSF2 (15%), EZH2 (14%), U2AF1, and RUNX1 (11% each), suggesting that mutations in these genes might precede the acquisition of other mutations. However, such statements are purely hypothesis generating in the absence of serial assessments of allele burden. On univariate analyses, significant associations were found between ASXL1 mutation and male sex, DNMT3A and IDH2 mutations and female sex, CBL and NRAS mutations and higher leukocyte count, NRAS mutation and lower hemoglobin level, SETBP1 and IDH2 mutations and higher BM blast percentage, and JAK2 mutation and longer diagnosis-to-referral interval.
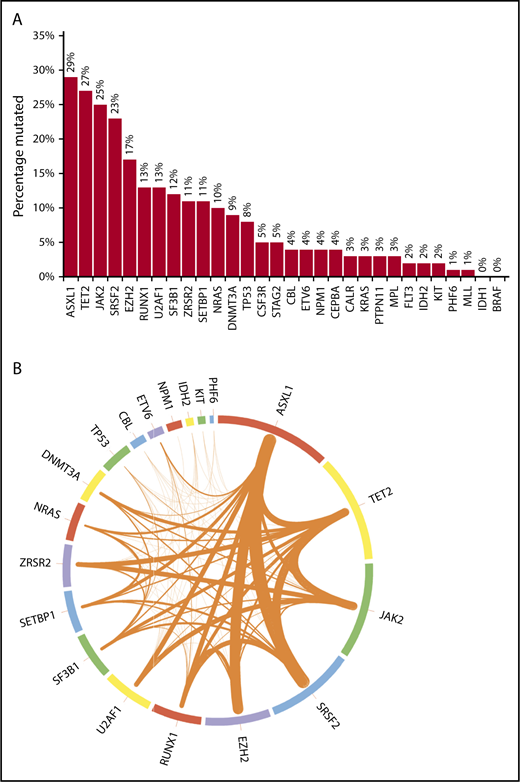
Bar chart and Circos plot of the results of targeted NGS in 102 patients with MDS/MPN-U. (A) Bar diagram showing the frequencies of selected gene mutations in the cohort of patients (see the third paragraph of text for the 20 genes sequenced in all patients). The other 10 genes of interest were sequenced in fewer patients, as follows: CALR in 58 (57%), MPL in 73 (72%), CEBPA in 78 (76%), KRAS in 86 (84%), BRAF and CSF3R in 57 (56%) each, FLT3 and PTPN11 in 87 (85%) each, and MLL and STAG2 in 77 (75%) each. One patient each had >1 mutation in ASXL1, CBL, SETBP1, U2AF1, and ZRSR2; 2 patients each had >1 mutation in DNMT3A, NRAS, and SF3B1; 3 had >1 mutation in EZH2; 4 had >1 mutation in TP53, and 5 had >1 mutation in TET2. (B) Circos plot depicting the mutational frequencies of the 20 genes tested in all 102 patients (length of the arcs), along with the frequencies of cooccurrence of different mutations in the same patient (width of the ribbons). The strongest relationships of cooccurrence were seen between ASXL1 and SRSF2 (n = 10), ASXL1 and EZH2 (n = 9), SRSF2 and TET2 (n = 8), TET2 and JAK2 (n = 8), and ASXL1 and TET2 (n = 7). Other readily evident patterns of comutation included ASXL1 with RUNX1 (n = 6), TET2 with ZRSR2 (n = 6), JAK2 with ZRSR2 (n = 6), TET2 with DNMT3A (n = 5), TET2 with SF3B1 (n = 5), and TET2 with RUNX1 (n = 5). Mutations in the signaling genes JAK2, CBL, and NRAS were mostly mutually exclusive. Only 1 patient each had JAK2/CBL and JAK2/NRAS comutation. CBL or NRAS mutations did not cooccur with CALR or MPL mutations. All JAK2 mutations were V617F and all CSF3R mutations T618I. All SRSF2 and IDH2 mutations involved the P95 and R140 residues, respectively. Most NRAS mutations affected G12 or G13, most SF3B1 mutations affected K700 or K666, and most U2AF1 mutations affected the Q157 residue.
Bar chart and Circos plot of the results of targeted NGS in 102 patients with MDS/MPN-U. (A) Bar diagram showing the frequencies of selected gene mutations in the cohort of patients (see the third paragraph of text for the 20 genes sequenced in all patients). The other 10 genes of interest were sequenced in fewer patients, as follows: CALR in 58 (57%), MPL in 73 (72%), CEBPA in 78 (76%), KRAS in 86 (84%), BRAF and CSF3R in 57 (56%) each, FLT3 and PTPN11 in 87 (85%) each, and MLL and STAG2 in 77 (75%) each. One patient each had >1 mutation in ASXL1, CBL, SETBP1, U2AF1, and ZRSR2; 2 patients each had >1 mutation in DNMT3A, NRAS, and SF3B1; 3 had >1 mutation in EZH2; 4 had >1 mutation in TP53, and 5 had >1 mutation in TET2. (B) Circos plot depicting the mutational frequencies of the 20 genes tested in all 102 patients (length of the arcs), along with the frequencies of cooccurrence of different mutations in the same patient (width of the ribbons). The strongest relationships of cooccurrence were seen between ASXL1 and SRSF2 (n = 10), ASXL1 and EZH2 (n = 9), SRSF2 and TET2 (n = 8), TET2 and JAK2 (n = 8), and ASXL1 and TET2 (n = 7). Other readily evident patterns of comutation included ASXL1 with RUNX1 (n = 6), TET2 with ZRSR2 (n = 6), JAK2 with ZRSR2 (n = 6), TET2 with DNMT3A (n = 5), TET2 with SF3B1 (n = 5), and TET2 with RUNX1 (n = 5). Mutations in the signaling genes JAK2, CBL, and NRAS were mostly mutually exclusive. Only 1 patient each had JAK2/CBL and JAK2/NRAS comutation. CBL or NRAS mutations did not cooccur with CALR or MPL mutations. All JAK2 mutations were V617F and all CSF3R mutations T618I. All SRSF2 and IDH2 mutations involved the P95 and R140 residues, respectively. Most NRAS mutations affected G12 or G13, most SF3B1 mutations affected K700 or K666, and most U2AF1 mutations affected the Q157 residue.
Median OS for the entire cohort was 12 months (from the sample date). Having ≥1 gene mutations among the 30 genes referred to in the previous paragraphs was associated with worse OS (median, 11.8 months with ≥1 mutations [93 patients] vs 28.6 months with no mutations [9 patients]; P = .024; supplemental Figure 1). When the analysis was limited to the 20 genes sequenced in all patients, the P value approached statistical significance (P = .077). Only CALR mutations (present in 2 of 58 patients tested) correlated significantly with improved survival (P = .02). Mutations in STAG2, CEBPA, and EZH2 were associated with trends toward inferior OS (P = .057, P = .09, and P = .08, respectively), and ZRSR2 mutations with a trend toward better OS (P = .09). At the last data cutoff point, 33 patients were alive; the median follow-up for these patients was 8 months (range, 1-173 months).
Sixteen patients (supplemental Table 3) developed disease transformation to acute myeloid leukemia (AML) a median of 10.8 months (range, 1-85 months) from the sample date. Patients with comutation of ASXL1 and SRSF2, each recognized as increasing the risk of leukemic transformation in patients with primary myelofibrosis (PMF),9 were significantly more likely to develop AML than those without ASXL1/SRSF2 comutation (4 of 10 vs 12 of 92; P = .026).
Comparison of our results with those of sequencing performed on 40 patients with chronic myelomonocytic leukemia (CMML) at the Cleveland Clinic (supplemental Table 4) revealed only the frequency of JAK2 mutations as being statistically significantly different, occurring in 25% of our patients but in only 1 patient (3%) in the CMML cohort (P = .003). Mutations in the CMML cohort also tended to occur at known hotspots.
Using the revised International Prognostic Scoring System (IPSS-R) for MDSs10 in all but 3 of our patients (with missing cytogenetic information), 13 (13%), 47 (46%), 19 (19%), 15 (15%), and 5 (5%) patients fell into the very low, low, intermediate-1, intermediate-2, and high-risk categories, respectively, with an overall P value of .07 for differences in OS. The Dynamic International Prognostic Scoring System (DIPSS) for primary myelofibrosis (PMF),11 however, did not stratify our patients into risk categories with significantly different survival; 7 (7%), 34 (33%), 39 (38%), and 22 (22%) of our patients were low, intermediate-1, intermediate-2, and high risk by the DIPSS, respectively, with an overall P value of .23 for between-group differences in OS.
As in MDS,12,13 the genes most frequently mutated in our cohort were mostly those encoding epigenetic regulators and splicing factors. There was a high frequency (24.5%) of JAK2 (V617F) mutations in our cohort, and very little cooccurrence with mutations in CBL or NRAS, other signaling genes. In 1 large study specifically looking at SETBP1, mutations in which have been considered a “biomarker” for MDS/MPN overlap syndromes,14 the mutational frequency among 253 patients with MDS/MPN-U was 12.3%, whereas it was 31.7% and 7.1% among those with aCML and CMML, respectively.15 SETBP1 mutations have been reported to confer a poor prognosis across MDS/MPN subtypes (aCML, MDS/MPN-U, CMML).15-17 SETBP1 mutations were detected at a frequency of 11% in our cohort, and no significant association was found with OS.
In an MDACC retrospective study of 85 patients with MDS/MPN-U, neither the IPSS-R for MDS10 nor the International Prognostic Scoring System (IPSS) for PMF18 was prognostically useful, whereas the original IPSS for MDS19 was.3 In the present study, which included patients at varying time points from diagnosis, we evaluated the IPSS-R for MDS10 and the DIPSS for PMF11 as both prognostic models have been validated at different time points in the respective disease courses. Our finding that the IPSS-R, but not the DIPSS, was prognostically informative in our patients with MDS/MPN-U, is interesting and requires further study.
Median survival in our cohort from time of referral was strikingly similar to that previously reported by the MDACC group (12.4 months),3 even though fewer than half of the patients in the current cohort were included in that report.3 In the prior study, no particular treatment regimen significantly improved response.3 Hypomethylating agents are often used,20 and the combination of ruxolitinib and azacitidine21 is emerging as an important treatment option. Although infrequent, mutations in genes such as IDH2 may afford opportunities for targeted therapy (eg, with enasidenib)22 in some patients, whether alone or in synergistic combinations.23
The online version of this article contains a data supplement.
Acknowledgments
This work was supported, in part, by the MD Anderson Cancer Center Support Grant P30 CA016672 from the National Institutes of Health, National Cancer Institute, the Charif Souki Cancer Research Fund (H.M.K.), and the Edward P. Evans Foundation (M.A.S. and J.P.M.).
Authorship
Contribution: P.B. analyzed data and wrote the paper; A.N., R.S.K., and M.R.S. collected data and critically reviewed the manuscript; S.L., C.B.-R., and A. Shaver reviewed pathology slides and confirmed the diagnoses; S.A.P. analyzed data; N.A.-A. and A. Sochacki collected data; K.P.P. and J.P.M. performed mutational analyses; W.M. and X.S. performed statistical analyses and provided the Circos plot; N.G.D., C.D.D., G.G.-M., H.M.K., M.A.S., and S.V. critically reviewed the manuscript; and S.V., J.P.M., R.S.K., and M.R.S. provided patient samples for mutational testing.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Prithviraj Bose, Department of Leukemia, University of Texas MD Anderson Cancer Center, 1400 Holcombe Blvd, FC4.3062, Unit 428, Houston, TX 77030; e-mail: pbose@mdanderson.org.
