

Key Points
Inactivation of Ikaros and Aiolos recapitulates the cell-intrinsic action of the IMiDs in MM, as well as transcriptional changes.
Loss of Ikaros or Aiolos results in upregulation of ISGs, including CD38, priming MM cells for anti-CD38 targeting.

Abstract
Recent studies have demonstrated that the immunomodulatory drugs (IMiDs) lead to the degradation of the transcription factors Ikaros and Aiolos. However, why their loss subsequently leads to multiple myeloma (MM) cell death remains unclear. Using CRISPR-Cas9 genome editing, we have deleted IKZF1/Ikaros and IKZF3/Aiolos in human MM cell lines to gain further insight into their downstream gene regulatory networks. Inactivation of either factor alone recapitulates the cell intrinsic action of the IMiDs, resulting in cell cycle arrest and induction of apoptosis. Furthermore, evaluation of the transcriptional changes resulting from their loss demonstrates striking overlap with lenalidomide treatment. This was not dependent on reduction of the IRF4-MYC “axis,” as neither protein was consistently downregulated, despite cell death occurring, and overexpression of either factor failed to rescue for Ikaros loss. Importantly, Ikaros and Aiolos repress the expression of interferon-stimulated genes (ISGs), including CD38, and their loss led to the activation of an interferon-like response, contributing to MM cell death. Ikaros/Aiolos repressed CD38 expression through interaction with the nucleosome remodeling and deacetylase complex in MM. IMiD-induced loss of Ikaros or treatment with interferon resulted in an upregulation of CD38 surface expression on MM cells, priming for daratumumab-induced NK cell-mediated antibody-dependent cellular cytotoxicity. These results give further insight into the mechanism of action of the IMiDs and provide mechanistic rationale for combination with anti-CD38 monoclonal antibodies.
Introduction
Multiple myeloma (MM) is an incurable cancer of postgerminal center plasma cells. The immunomodulatory drugs (IMiDs), including thalidomide, lenalidomide, and pomalidomide, are a cornerstone of current MM therapy. Our understanding of the underlying mechanism of action of the IMiDs has advanced considerably in recent years with the identification that thalidomide directly binds to cereblon, the substrate receptor of an E3 ubiquitin ligase complex.1 On IMiD binding, cereblon acquires the ability to ubiquitinate and degrade novel substrates, including the related transcription factors Ikaros (IKZF1) and Aiolos (IKZF3), resulting in MM cell death.2-4 How loss of either or both of these transcription factors results in myeloma cell death is unclear.
Ikaros is vital for B-cell lymphopoiesis, and Ikaros null mice lack B cells from the earliest stage of differentiation.5-7 Ikaros plays numerous important roles in B-cell development, including Igh rearrangement and, together with Aiolos, regulates the pre-B-cell transition.8 At this time, the role of Ikaros in normal plasma cells is unknown. Aiolos has been demonstrated to be important for the production of high-affinity long-lived bone marrow plasma cells.9
The action of the IMiDs has been linked to a reduction in the transcription factor interferon (IFN) regulatory factor 4 (IRF4). IRF4 is essential for the development and survival of normal plasma cells, with its loss resulting in cell death in MM10-13 and activated B cell–subtype diffuse large B-cell lymphoma (DLBCL); however, not germinal center B cell–subtype DLBCL.14 Lenalidomide has a similar spectrum of disease activity and was subsequently shown to also result in overlapping gene expression changes with IRF4 targeting.14 Although cell death resulting from IRF4 loss remains poorly understood, MYC has been identified as an important downstream IRF4 target. It is currently a widely accepted model that Ikaros and Aiolos are upstream activators of IRF4, and hence MYC; however, this has not been thoroughly investigated.
Two recent studies have suggested alternative or additional actions of the IMiDs that are largely independent of Ikaros and Aiolos. The IMiDs were reported to impair cereblon’s binding and stabilization of the CD147–MCT1 complex, leading to MM cytotoxicity,15 whereas a second study showed cereblon-dependent inhibition of thioredoxin reductase, resulting in increased intracellular H2O2, accumulation of immunoglobulin light-chain dimers, and endoplasmic reticulum–stress induced cytotoxicity.16 Although Ikaros and Aiolos degradation was found to be a consequence of increased H2O2 in the latter study, their loss was not reported to be essential for the action of the IMiDs.
In this study, we sought to further understand the biological role of Ikaros and Aiolos in MM cells, and specifically to determine how their targeting contributes to the action of the IMiDs. We find that loss of either Ikaros or Aiolos alone recapitulates the molecular changes associated with IMiD exposure. Ikaros and Aiolos repressed a swathe of IFN-regulated genes, including the gene encoding CD38, the target of the monoclonal antibody (mAb) daratumumab, providing a clear molecular explanation for the clinical benefit of combinatorial treatment of MM with lenalidomide and daratumumab.
Methods
Cell culture
OPM2 (DSMZ), H929 (ATCC), MM1S (ATCC), RPMI-8226 (ATCC), U266B1 (ATCC), and AMO-1 (DSMZ) MM cell lines were from David Huang (The Walter and Eliza Hall Institute).17 Bone marrow samples were acquired from patients with newly diagnosed (treatment-naive) MM after written informed consent. MM cells were isolated by Ficoll separation followed by flow cytometric sorting of CD38hiCD138hi cells. NK cells were isolated from the blood of healthy donors by Ficoll separation followed by immunomagnetic negative selection (Stemcell Technologies, Vancouver, Canada). Cell culture, drug treatment, and cytotoxicity assays are described in the supplemental Methods, available on the Blood Web site. Access to donor samples was approved by the Monash Health and Walter and Eliza Hall Institute human research ethics committees.
CRISPR-Cas9-mediated genome editing
MM cell lines were sequentially transduced with lentivirus for a constitutive Cas9 expression vector tagged with mCherry, and then doxycycline-inducible guide-RNA (gRNA) vectors were tagged with either green fluorescent protein (GFP) or cyan fluorescent protein (CFP).18 Cells were then sorted by flow cytometry.
The DNA sequences of the gRNAs targeting IKZF1/Ikaros, Ik19, Ik20, and IKZF3/Aiolos, Ai35 and Ai38, are shown on Figure 1A,D. Expression of the gRNAs was induced by doxycycline (1 μg/mL), and genome editing confirmed by western blotting and DNA sequencing.
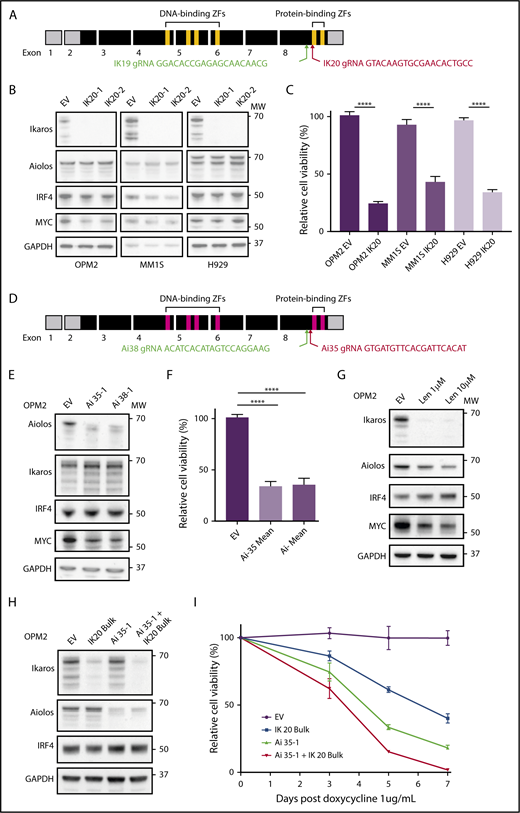
CRISPR-Cas9 deletion of IKZF1/3 recapitulates the action of the IMiDs in MM cells. (A) Depiction of full-length IKZF1 gene and CRISPR targeting strategy. (B) Western blot of Cas9 expressing OPM2, MM1S, and H929 clones of IKZF1 gRNA IK20 or an empty vector (EV) control performed 4 days after gRNA induction with doxycycline (dox). Membranes were probed for Ikaros, Aiolos, IRF4, MYC, and GAPDH as a loading control. (C) CellTiter Glo (CTG) viability assay 6 days after dox treatment of cells (as in panel B). Relative cell viability vs non-dox-treated control (set to 100%) is shown. Data are the mean ± SD from 3 experiments. (D) Depiction of full-length IKZF3 gene and CRISPR targeting strategy. (E) Western blot of Cas9-expressing OPM2 clones of each IKZF3 gRNA (Ai35 and Ai38) vs EV 4 days after dox treatment. Membranes were probed for Aiolos, Ikaros, IRF4, MYC, and GAPDH as a loading control. (F) CTG viability assay 6 days after dox treatment of OPM2 clones (as in panel E). Relative cell viability vs non-dox-treated control (set to 100%) is shown. Data are the mean ± SD from 3 experiments. (G) Western blot of OPM2 cells 2 days posttreatment with lenalidomide (Len, 1 or 10 μM) vs control, probed for Ikaros, Aiolos, MYC, IRF4, and GAPDH as a loading control. (H) Western blot of Cas9 expressing OPM2 cells (lanes 1 and 2) or IKZF3 gRNA Ai35-1 clone (lanes 3 and 4), transduced with IKZF1 IK20 gRNA (lanes 2 and 4) or EV control (lane 1) and bulk flow cytometry sorted for mCherry (Cas9), GFP (IK20 gRNA) expression. Analysis was performed 72 hours after dox treatment to induce gRNA expression. Membrane were probed for Ikaros, Aiolos, IRF4, and GAPDH as a loading control. (I) Flow cytometry viability time course (using a fixable viability dye and counting beads to quantify live cells) after dox treatment in OPM2 IK20 (bulk), Ai35-1 (clone), combination IK20 + Ai35-1, or EV control cells (as in panel H). Relative cell viability vs non-dox-treated control (set to 100%) is shown. Data are the mean ± SD from 3 experiments. (B,E,G,H) Molecular weights (MW) are indicated to the right of the plots. ****P < .0001, using an unpaired Student t test.
CRISPR-Cas9 deletion of IKZF1/3 recapitulates the action of the IMiDs in MM cells. (A) Depiction of full-length IKZF1 gene and CRISPR targeting strategy. (B) Western blot of Cas9 expressing OPM2, MM1S, and H929 clones of IKZF1 gRNA IK20 or an empty vector (EV) control performed 4 days after gRNA induction with doxycycline (dox). Membranes were probed for Ikaros, Aiolos, IRF4, MYC, and GAPDH as a loading control. (C) CellTiter Glo (CTG) viability assay 6 days after dox treatment of cells (as in panel B). Relative cell viability vs non-dox-treated control (set to 100%) is shown. Data are the mean ± SD from 3 experiments. (D) Depiction of full-length IKZF3 gene and CRISPR targeting strategy. (E) Western blot of Cas9-expressing OPM2 clones of each IKZF3 gRNA (Ai35 and Ai38) vs EV 4 days after dox treatment. Membranes were probed for Aiolos, Ikaros, IRF4, MYC, and GAPDH as a loading control. (F) CTG viability assay 6 days after dox treatment of OPM2 clones (as in panel E). Relative cell viability vs non-dox-treated control (set to 100%) is shown. Data are the mean ± SD from 3 experiments. (G) Western blot of OPM2 cells 2 days posttreatment with lenalidomide (Len, 1 or 10 μM) vs control, probed for Ikaros, Aiolos, MYC, IRF4, and GAPDH as a loading control. (H) Western blot of Cas9 expressing OPM2 cells (lanes 1 and 2) or IKZF3 gRNA Ai35-1 clone (lanes 3 and 4), transduced with IKZF1 IK20 gRNA (lanes 2 and 4) or EV control (lane 1) and bulk flow cytometry sorted for mCherry (Cas9), GFP (IK20 gRNA) expression. Analysis was performed 72 hours after dox treatment to induce gRNA expression. Membrane were probed for Ikaros, Aiolos, IRF4, and GAPDH as a loading control. (I) Flow cytometry viability time course (using a fixable viability dye and counting beads to quantify live cells) after dox treatment in OPM2 IK20 (bulk), Ai35-1 (clone), combination IK20 + Ai35-1, or EV control cells (as in panel H). Relative cell viability vs non-dox-treated control (set to 100%) is shown. Data are the mean ± SD from 3 experiments. (B,E,G,H) Molecular weights (MW) are indicated to the right of the plots. ****P < .0001, using an unpaired Student t test.
Protein characterization
Protocols and antibodies used for western blotting, coimmunoprecipitation, and flow cytometry are described in the supplemental Methods.
RNA sequencing
After doxycycline treatment of the stated duration, live mCherry+ (Cas9), CFP/GFP+ (gRNA) cells were sorted by flow cytometry. RNA extraction was then performed using the Qiagen RNeasy Plus Kit. RNA library preparation was conducted using the Illumina Truseq Kit. Sequencing was performed on an Illumina NextSeq sequencer with 80-bp single-end reads achieving an average of 2.2 × 107 reads/sample. Technical duplicates were sequenced for each time for empty vector controls and at least biological duplicates for IKZF1/IKZF3 gRNA clones in each cell line. Reads were aligned to the GRCh38/hg38 build of the human genome and analyzed, as described in the supplemental Methods.
Statistical analysis
Statistical significance of non-RNAseq data were assessed with Prism6. Student t tests were unpaired, assumed Gaussian distribution and that both populations have the same standard deviation (SD). Bar graphs display the arithmetic mean ± SD.
Accession codes
Raw sequence reads, read counts, and normalized expression values have been deposited into the Gene Expression Omnibus database under accession number GSE113031.
Results
CRISPR-Cas9 inactivation of IKZF1/3 in MM cells
The IKZF1 gene encodes multiple isoforms produced through alternate splicing. Isoforms containing fewer than 3 of the 4 N-terminal zinc fingers (encoded by exons 4-6; Figure 1A) are unable to bind DNA efficiently; however, they continue to interact with other Ikaros isoforms through the C-terminal zinc fingers and have dominant negative function.19 To target all Ikaros isoforms and avoid the production of a dominant-negative protein, we designed gRNAs targeting exon 8 upstream of the C-terminal zinc fingers (Figure 1A).6,20 This strategy is similar to that used previously in murine models to produce a null allele.6,19
Two gRNAs (IK19 and IK20) were designed and cloned into the doxycycline inducible fgh1t lentiviral vector,18 which is tagged with either GFP or CFP. Three IMiD-sensitive human myeloma cell lines, OPM2, MM1S, and H929, were transduced with a constitutive Cas9 expression vector tagged with mCherry, and each of the IKZF1 gRNAs lentivirus, and sorted by flow cytometry for mCherry and GFP (or CFP) expression. Treatment with doxycycline resulted in efficient deletion of Ikaros in cells transduced with either gRNA (supplemental Figure 1A-B). IK20 lead to more efficient knockdown of Ikaros and was used for the majority of subsequent experiments (supplemental Figure 1A-D).
Ikaros or Aiolos loss recapitulates the MM cell-intrinsic action of the IMiDs
Consistent with its reported role in the action of the IMiDs, inactivation of Ikaros resulted in reduced cell viability in all 3 cell lines (Figure 1B-C).21,22 This was through both the induction of apoptosis, as demonstrated by cell viability studies, and increased expression of cleaved caspase 3 (supplemental Figure 1C,E), as well as a G0/1 cell cycle arrest (supplemental Figure 1F-G). Further recapitulating the action of the IMiDs in MM, potent synergy was seen with loss of Ikaros in combination with low-dose dexamethasone treatment (supplemental Figure 1H).22
gRNAs were also designed using a similar strategy to target IKZF3/Aiolos (Figure 1D). Loss of Aiolos in OPM2 cells resulted in marked loss of viability, with similar timing and efficiency as seen with Ikaros (Figure 1E-F,H,I). Inactivation of Ikaros or Aiolos did not affect the other’s expression (Figure 1B,E), suggesting that Ikaros and Aiolos function in a largely nonredundant capacity in MM. We did, however, observe a small degree of functional compensation between the 2 proteins as the combined inactivation of Ikaros and Aiolos, through bulk (ie, less efficient) IK20 gRNA transduction of the Aiolos 35-1 clone and subsequent doxycycline treatment to induce both gRNA, resulted in slightly augmented cell death (Figure 1H-I).
Cell death induced by Ikaros inactivation is not dependent on reduced IRF4 and MYC
An IRF4-MYC axis has been proposed to be the critical downstream target of Ikaros/Aiolos degradation secondary to the IMiDs. However, despite Ikaros being required for viability in all 3 cell lines, a heterogenous effect on IRF4 and MYC expression was observed (Figure 1B-C). In MM1S cells, Ikaros deficiency resulted in reduced IRF4 and a small reduction in MYC expression, consistent with current models. In contrast, in OPM2 cells, loss of either Ikaros or Aiolos, combined inactivation of both Ikaros and Aiolos, or treatment with lenalidomide resulted in a marked downregulation of MYC, with no change in IRF4 (Figure 1B,E,G-H). Conversely, neither IRF4 nor MYC was dependent on Ikaros in H929 cells, despite cell death also occurring. These results suggest that that the loss of viability induced on inactivation of Ikaros, or treatment with the IMiDs, is not universally dependent on the reduction of either IRF4 or MYC, although they are likely to be important additional factors in certain tumor genotypes.
MYC or IRF4 overexpression fails to complement Ikaros deficiency
As loss of MYC and IRF4 have individually been demonstrated to result in MM cell death,10 we sought to determine to what extent downregulation of each factor explains the cell death resulting from loss of Ikaros. To examine this, MYC was overexpressed in Ikaros-deficient MM cells. In OPM2 cells in which Ikaros deficiency resulted in a reduction of endogenous MYC, retroviral overexpression led to a restoration of MYC protein expression approximating baseline (Figure 2A). Enforced MYC expression initially resulted in a partial rescue of viable cell numbers after Ikaros inactivation; however, this effect was short-lived, with cell death ensuing despite MYC expression (Figure 2B). In contrast, MYC overexpression failed to any extent to rescue Ikaros loss in MM1S cells (Figure 2C-D).
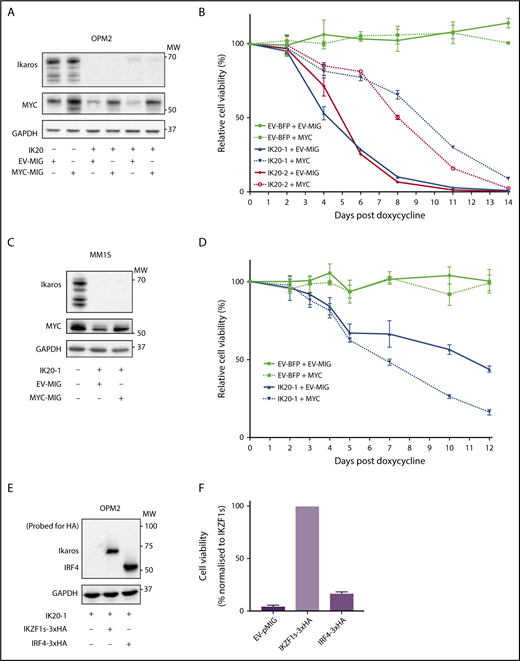
MYC or IRF4 overexpression fails to rescue for loss of Ikaros. (A) Western blot of OPM2 IKZF1 gRNA clones IK20-1 and IK20-2 or EV cells transduced with either MYC MSCV-IRES-GFP (MYC-MIG) or EV-MIG control 4 days postdox treatment. Membranes were probed for Ikaros, MYC, and GAPDH. (B) CTG cell viability time course of samples described in (A) at the indicated days after treatment with dox. Data are the mean ± SD from 3 experiments. (C) Western blot of MM1S IKZF1 gRNA clone IK20-1 or EV cells transduced with either MYC MSCV-IRES-GFP (MYC-MIG) or EV-MIG control 4 days postdox. Membranes were probed for Ikaros, MYC, and GAPDH. (D) Cell viability time course of samples described in (C) after treatment with dox. Data are the mean ± SD from 3 experiments. (E) Western blot of OPM2 IKZF1 gRNA clone IK20-1 transduced with either HA-tagged full-length IRF4 MIG (IRF4-3xHA), an EV-MIG negative control, or a CRISPR-resistant IKZF1 construct (IKZF1s-3xHA). Membranes were probed with anti-HA and GAPDH 3 days postdox treatment. (A,C,E) MW are indicated to the right of the plots, GAPDH is a loading control. (F) CTG viability assay of samples described in panel E, 10 days after dox treatment. Relative cell viability vs non-dox-treated control normalized to IKZF1s-3xHA (set to 100%) is shown. Data are the mean ± SD from 3 experiments.
MYC or IRF4 overexpression fails to rescue for loss of Ikaros. (A) Western blot of OPM2 IKZF1 gRNA clones IK20-1 and IK20-2 or EV cells transduced with either MYC MSCV-IRES-GFP (MYC-MIG) or EV-MIG control 4 days postdox treatment. Membranes were probed for Ikaros, MYC, and GAPDH. (B) CTG cell viability time course of samples described in (A) at the indicated days after treatment with dox. Data are the mean ± SD from 3 experiments. (C) Western blot of MM1S IKZF1 gRNA clone IK20-1 or EV cells transduced with either MYC MSCV-IRES-GFP (MYC-MIG) or EV-MIG control 4 days postdox. Membranes were probed for Ikaros, MYC, and GAPDH. (D) Cell viability time course of samples described in (C) after treatment with dox. Data are the mean ± SD from 3 experiments. (E) Western blot of OPM2 IKZF1 gRNA clone IK20-1 transduced with either HA-tagged full-length IRF4 MIG (IRF4-3xHA), an EV-MIG negative control, or a CRISPR-resistant IKZF1 construct (IKZF1s-3xHA). Membranes were probed with anti-HA and GAPDH 3 days postdox treatment. (A,C,E) MW are indicated to the right of the plots, GAPDH is a loading control. (F) CTG viability assay of samples described in panel E, 10 days after dox treatment. Relative cell viability vs non-dox-treated control normalized to IKZF1s-3xHA (set to 100%) is shown. Data are the mean ± SD from 3 experiments.
To evaluate the role of IRF4 as a critical downstream target of Ikaros, we overexpressed hemagglutinin-tagged IRF4 or a CRISPR-resistant hemagglutinin-tagged full-length IKZF1 ORF (IKZF1s) as a positive control in Ikaros-deficient OPM2 or H929 cells. In contrast to IKZF1s, enforced IRF4 expression failed to rescue cell viability on Ikaros inactivation in either cell line (Figure 2E-F; supplemental Figure 2A-B). Collectively, these results strongly support the central role of Ikaros and Aiolos in the MM cell-intrinsic action of the IMiDs; however, the loss of cell viability cannot be solely explained by a deficiency in the IRF4-MYC axis.
Investigating the transcriptional changes on loss of Ikaros, Aiolos, or lenalidomide treatment
RNA sequencing was performed to further investigate the transcriptional changes induced by the loss of Ikaros in OPM2, MM1S, and H929 cells (supplemental Figure 3A; supplemental Tables 1-5). Mapped reads were also analyzed to confirm efficient IKZF1 targeting (supplemental Figure 3B). In OPM2 cells, significant overlap was seen in differential gene expression after deletion of Ikaros and Aiolos, with 747 common upregulated (repressed by Ikaros/Aiolos) targets, accounting for 68% and 64% of total Ikaros and Aiolos upregulated genes, respectively, and 857 common downregulated targets (required Ikaros/Aiolos for their expression), accounting for 65% and 69% of total Ikaros and Aiolos downregulated genes, respectively (Figure 3A). This further supports the hypothesis that Ikaros and Aiolos function in a similar, however nonredundant, fashion in MM. The combination of Ikaros and Aiolos loss accounted for 74% of upregulated and 50% of downregulated genes after lenalidomide treatment (Figure 3A), strongly supporting their central role in IMiD function.
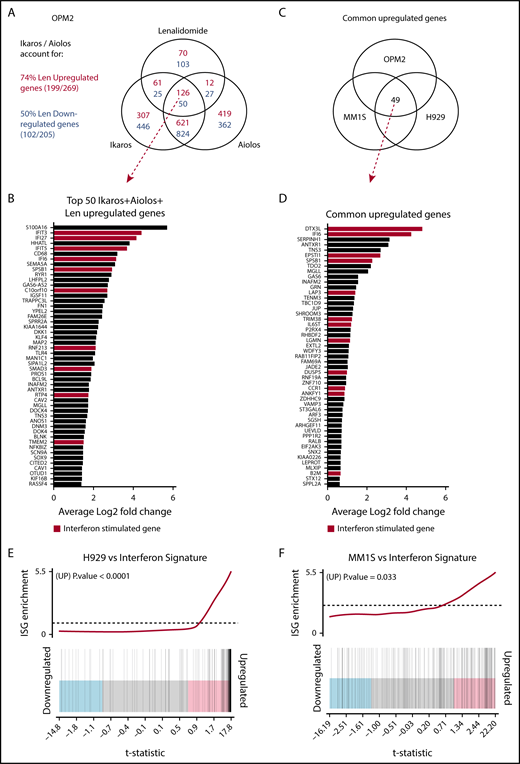
Investigating the transcriptional changes resulting from loss of Ikaros, Aiolos, or lenalidomide treatment. RNA sequencing (RNAseq) was performed in OPM2, MM1S, and H929 cells 72 hours after IKZF1 (IK20) gRNA induction vs EV control. In OPM2 cells RNAseq was also performed after IKZF3 (Ai35) gRNA induction with dox or treatment with 10 μM lenalidomide. A cutoff false-discovery rate of 0.15 and additional criteria of a fold change ≥1.5 and an RPKM (reads per kilobase of exon length per million mapped fragments) ≥1 were employed for calling of differentially expressed genes. (A) Venn diagram depicting overlap of differentially expressed genes after inactivation of Ikaros, Aiolos, or treatment with lenalidomide in OPM2 cells. Upregulated (Ikaros/Aiolos repressed) genes are in red, downregulated (Ikaros/Aiolos activated) genes are in blue. (B) Top 50 shared upregulated genes in OPM2 cells after loss of Ikaros, Aiolos, or lenalidomide treatment. ISGs are highlighted in red. (C) Venn diagram and (D) graph depicting overlap of common upregulated genes on deletion of Ikaros in OPM2, MM1S, and H929 cells. ISGs are highlighted in red. (E-F) Gene set enrichment analysis and barcode plot of differential gene expression in IK20 in (E) H929 and (F) MM1S cells after dox treatment (as in panel A), tested against a curated list of ISGs from published sources (list shown in supplemental Table 6).14,24,25 The differential gene expression data set is shown as a shaded rectangle with genes horizontally ranked by moderated t statistic. Genes upregulated upon Ikaros loss are shaded pink and downregulated genes shaded blue. The position of individual ISGs is marked on the plot by vertical black lines. P values for the gene set enrichment test are shown above.
Investigating the transcriptional changes resulting from loss of Ikaros, Aiolos, or lenalidomide treatment. RNA sequencing (RNAseq) was performed in OPM2, MM1S, and H929 cells 72 hours after IKZF1 (IK20) gRNA induction vs EV control. In OPM2 cells RNAseq was also performed after IKZF3 (Ai35) gRNA induction with dox or treatment with 10 μM lenalidomide. A cutoff false-discovery rate of 0.15 and additional criteria of a fold change ≥1.5 and an RPKM (reads per kilobase of exon length per million mapped fragments) ≥1 were employed for calling of differentially expressed genes. (A) Venn diagram depicting overlap of differentially expressed genes after inactivation of Ikaros, Aiolos, or treatment with lenalidomide in OPM2 cells. Upregulated (Ikaros/Aiolos repressed) genes are in red, downregulated (Ikaros/Aiolos activated) genes are in blue. (B) Top 50 shared upregulated genes in OPM2 cells after loss of Ikaros, Aiolos, or lenalidomide treatment. ISGs are highlighted in red. (C) Venn diagram and (D) graph depicting overlap of common upregulated genes on deletion of Ikaros in OPM2, MM1S, and H929 cells. ISGs are highlighted in red. (E-F) Gene set enrichment analysis and barcode plot of differential gene expression in IK20 in (E) H929 and (F) MM1S cells after dox treatment (as in panel A), tested against a curated list of ISGs from published sources (list shown in supplemental Table 6).14,24,25 The differential gene expression data set is shown as a shaded rectangle with genes horizontally ranked by moderated t statistic. Genes upregulated upon Ikaros loss are shaded pink and downregulated genes shaded blue. The position of individual ISGs is marked on the plot by vertical black lines. P values for the gene set enrichment test are shown above.
Loss of Ikaros, Aiolos, or lenalidomide treatment increases expression of IFN-stimulated genes
Next we compared the transcriptional changes on loss of Ikaros across the 3 cell lines (Figure 3B). Analysis by multidimensional scaling plot demonstrated striking heterogeneity of steady-state gene expression between the cell lines, in keeping with their diverse chromosomal, mutational, and epigenetic landscapes (supplemental Figure 3C). Despite this heterogeneity, upregulation of numerous IFN-stimulated genes (ISGs) was observed in all 3 cell lines after deletion of Ikaros, as well as loss of Aiolos and lenalidomide treatment in OPM2 cells (Figure 3A-D; supplemental Figure 3D).23 This was confirmed by gene set enrichment analysis of a curated list of ISGs from published sources (Figure 3E-F).14,24,25 This increased expression was particularly striking in H929 cells (P < .0001), where ISGs constituted 29 of the top 50 upregulated genes (supplemental Figure 3D) and also reached statistical significance in MM1S cells (P = .033). In OPM2 cells, this was close to reaching statistical significance for Ikaros (P = .074) or Aiolos (P = .063; supplemental Figure 3E-F). Upregulation of ISGs was not the result of an increased expression of any IFN genes or subunits of the IFN type I or II receptors (data not shown). A role of Aiolos in the repression of ISGs has recently been described in DLBCL.26 In keeping with this, significant overlap was seen after loss of Ikaros in these MM cell lines with published gene expression changes resulting from treatment with CC-122, an IMiD-like compound that results in cereblon-dependent degradation of Ikaros/Aiolos, in the DLBCL line OCI-Ly1026 (supplemental Figure 4).
Assessing the importance of ISGs for IMiD function
We next sought to validate the upregulation of ISGs at a protein level. In agreement with the RNAseq data (Figure 4A), a marked increased expression of interferon-induced protein with tetratricopeptide repeats (IFIT3) was seen on loss of Ikaros by western blotting (Figure 4B). Analysis of Ikaros ChIPseq data from the EBV transformed B-cell line GM1287827 demonstrated Ikaros binding peaks in the 2 promoter regions of IFIT3, suggesting direct repression of this gene (Figure 4C).
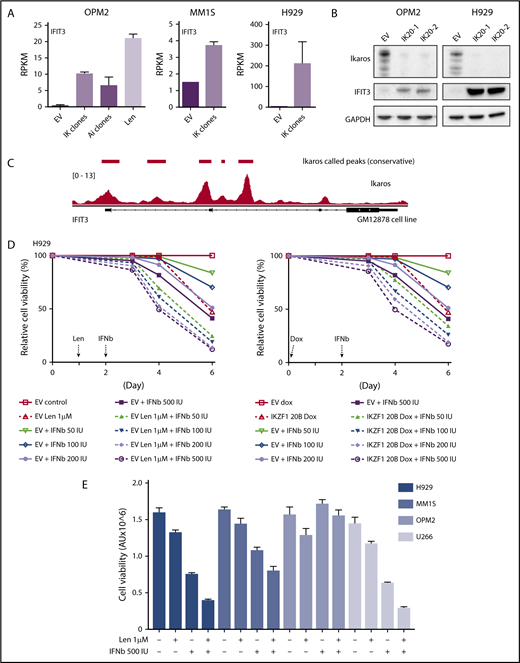
Assessing the importance of interferon-stimulated gene upregulation in the action of the IMiDs. (A) IFIT3 expression (mean RPKM ± SD) from RNA sequencing in OPM2 cell after deletion of Ikaros, Aiolos, or lenalidomide (Len) 10 μM; and MM1S and H929 cells after deletion of Ikaros, vs control. Data are described in Figure 3. (B) Western blot OPM2 and H929 clones of IKZF1 20 (IK20) gRNA (2 clones) and an EV control, performed 4 days after gRNA induction with dox. Membranes were probed for Ikaros, IFIT3, and GAPDH as loading control. (C) Analysis of Ikaros binding in the human B-cell line GM12878 by ChIP-seq from the ENCODE Project. Shown is the mapping track of sequence reads at the IFIT3 locus. Note the presence of 2 alternative first IFIT3 exons. Ikaros conservative called peaks are shown as bars in the top panel. (D) Time course of cell viability assayed by flow cytometry using a fixable viability dye. Relative viability vs non-dox-treated control (set to 100%) is shown. Data are representative of 2 experiments. Graph shows live cell numbers in H929 cells expressing the control EV or IKZF1 gRNA (IK20) after dox gRNA induction (day 0), Len 1 μM treatment (day 1), and/or a titration of β-IFN (day 2). IU, international units. (E) CellTiter Glo viability assay in H929, MM1S, OPM2, and U266 cells 3 days posttreatment with Len 1 μM and/or β-IFN 500 IU. Graph depicts the mean luminescent reading from the Cell Titer Glo assay expressed as an arbitrary unit (AU) ± SD for each sample from 3 experiments.
Assessing the importance of interferon-stimulated gene upregulation in the action of the IMiDs. (A) IFIT3 expression (mean RPKM ± SD) from RNA sequencing in OPM2 cell after deletion of Ikaros, Aiolos, or lenalidomide (Len) 10 μM; and MM1S and H929 cells after deletion of Ikaros, vs control. Data are described in Figure 3. (B) Western blot OPM2 and H929 clones of IKZF1 20 (IK20) gRNA (2 clones) and an EV control, performed 4 days after gRNA induction with dox. Membranes were probed for Ikaros, IFIT3, and GAPDH as loading control. (C) Analysis of Ikaros binding in the human B-cell line GM12878 by ChIP-seq from the ENCODE Project. Shown is the mapping track of sequence reads at the IFIT3 locus. Note the presence of 2 alternative first IFIT3 exons. Ikaros conservative called peaks are shown as bars in the top panel. (D) Time course of cell viability assayed by flow cytometry using a fixable viability dye. Relative viability vs non-dox-treated control (set to 100%) is shown. Data are representative of 2 experiments. Graph shows live cell numbers in H929 cells expressing the control EV or IKZF1 gRNA (IK20) after dox gRNA induction (day 0), Len 1 μM treatment (day 1), and/or a titration of β-IFN (day 2). IU, international units. (E) CellTiter Glo viability assay in H929, MM1S, OPM2, and U266 cells 3 days posttreatment with Len 1 μM and/or β-IFN 500 IU. Graph depicts the mean luminescent reading from the Cell Titer Glo assay expressed as an arbitrary unit (AU) ± SD for each sample from 3 experiments.
α-IFN treatment has previously been trialed in patients with MM with varying efficacy.28-33 Preclinical studies have also previously demonstrated heterogeneity of response of myeloma cell lines to IFN.34-37 Given this, we sought to clarify whether this increased expression of ISGs contributes to cell death in these cell lines. As this effect was most profound in H929 cells, we initially focused on this cell line. Titration of β-IFN led to a dose-dependent loss of viability, indicating that these cells are indeed sensitive to perturbation of the β-IFN pathway. Furthermore, an additive loss of cell viability was demonstrated when β-IFN was combined with either deletion of Ikaros or treatment with lenalidomide (Figure 4D). A similar additive upregulation of IFIT3 expression occurred after combination treatment with β-IFN and lenalidomide or Ikaros inactivation (supplemental Figure 5). These results suggest that upregulation of ISGs is likely to be an important factor contributing to cell death in this cell line upon lenalidomide-induced Ikaros/Aiolos degradation. An additive loss of cell viability was also seen with the combination of lenalidomide and β-IFN in U266 and MM1S cells, but not OPM2 cells, for unknown reasons (Figure 4E).
The IFN-regulated gene CD38 is repressed by Ikaros and Aiolos
CD38 is a surface glycoprotein that is uniformly expressed in normal plasma cells and in the majority of MM.38,39 CD38 mRNA expression was increased on loss of Ikaros, Aiolos, or lenalidomide treatment in OPM2 cells and after Ikaros deletion in H929 cells and MM1S cells (Figure 5A). Consistent with these results, treatment with lenalidomide increased surface expression of CD38 in H929, OPM2, and AMO-1 cells (Figure 5B; supplemental Figure 6A). Interestingly, no increase was seen in MM1S cells despite increased RNA expression, suggesting additional posttranslational control of expression. As CD38 has been documented to be an IFN regulated in a number of normal and malignant hematopoietic cell types,40-42 we next sought to define whether CD38 is indeed an ISG in the context of MM. In both H929 and OPM2 cells, deletion of Ikaros or treatment with low-dose β-IFN led to increased surface expression of CD38, and the combination of Ikaros loss and β-IFN or lenalidomide and β-IFN resulted in an additive increase in expression (Figures 5C and 6D; supplemental Figure 6B). Finally, analysis of 5 treatment-naive MM samples also demonstrated increased CD38 expression after lenalidomide or β-IFN treatment, and again an additive effect with the combination (Figure 5D; supplemental Figure 6C).

CD38 upregulation through lenalidomide-induced loss of Ikaros or interferon treatment enhances daratumumab-stimulated NK-cell mediated ADCC. (A) CD38 expression (mean RPKM ± SD) from RNA sequencing in OPM2 cells after deletion of Ikaros, Aiolos, or lenalidomide (Len) 10 μM, and MM1S and H929 cells after deletion of Ikaros, vs control. Data are described in Figure 3. (B) CD38 mean fluorescence intensity (MFI) ± SD determined (relative to control) by flow cytometry from 3 experiments in H929, OPM2, AMO-1, and MM1S cells 48 hours after 0, 1, or 10 μM Len. ****P < .0001 using an unpaired Student t test. Refer to supplemental Figure 6 for corresponding flow cytometry histograms. (C) Histogram of CD38 surface expression by flow cytometry in H929 cells at baseline, or 96 hours after dox IKZF1 gRNA induction, 72 hours after treatment with 1μ M Len, and/or 48 hours after 50 IU β-IFN. (D) CD38 MFI ± SD determined by flow cytometry in 5 newly diagnosed treatment-naïve MM patient samples isolated from bone marrow aspirate by flow cytometry sorting and treated for 60-72 hours with 1 μM Len, 100 IU β-IFN, combination Len + β-IFN, or control. ***P < .001 using an unpaired Student t test. (E) Calcein-AM cell lysis assay evaluating human NK cell (effector)-mediated antibody-dependent cellular cytotoxicity of H929 (target) cells after a dose titration of daratumumab. Effector to target (E:T) ratio 5:1. H929 IKZF1 gRNA cells pretreated with dox (day, −4), lenalidomide 1 μM (day −3), and/or β-IFN 50 IU (day −2) before assay (D0). Percentage lysis determined relative to internal “spontaneous” (no NK cells or daratumumab) and “max” (media with 2% Triton-X) lysis controls for each sample to exclude effect of differing pretreatments. Data are the mean % lysis ± SD and are representative of 4 experiments. The untreated H929 control data are common to both data sets.
CD38 upregulation through lenalidomide-induced loss of Ikaros or interferon treatment enhances daratumumab-stimulated NK-cell mediated ADCC. (A) CD38 expression (mean RPKM ± SD) from RNA sequencing in OPM2 cells after deletion of Ikaros, Aiolos, or lenalidomide (Len) 10 μM, and MM1S and H929 cells after deletion of Ikaros, vs control. Data are described in Figure 3. (B) CD38 mean fluorescence intensity (MFI) ± SD determined (relative to control) by flow cytometry from 3 experiments in H929, OPM2, AMO-1, and MM1S cells 48 hours after 0, 1, or 10 μM Len. ****P < .0001 using an unpaired Student t test. Refer to supplemental Figure 6 for corresponding flow cytometry histograms. (C) Histogram of CD38 surface expression by flow cytometry in H929 cells at baseline, or 96 hours after dox IKZF1 gRNA induction, 72 hours after treatment with 1μ M Len, and/or 48 hours after 50 IU β-IFN. (D) CD38 MFI ± SD determined by flow cytometry in 5 newly diagnosed treatment-naïve MM patient samples isolated from bone marrow aspirate by flow cytometry sorting and treated for 60-72 hours with 1 μM Len, 100 IU β-IFN, combination Len + β-IFN, or control. ***P < .001 using an unpaired Student t test. (E) Calcein-AM cell lysis assay evaluating human NK cell (effector)-mediated antibody-dependent cellular cytotoxicity of H929 (target) cells after a dose titration of daratumumab. Effector to target (E:T) ratio 5:1. H929 IKZF1 gRNA cells pretreated with dox (day, −4), lenalidomide 1 μM (day −3), and/or β-IFN 50 IU (day −2) before assay (D0). Percentage lysis determined relative to internal “spontaneous” (no NK cells or daratumumab) and “max” (media with 2% Triton-X) lysis controls for each sample to exclude effect of differing pretreatments. Data are the mean % lysis ± SD and are representative of 4 experiments. The untreated H929 control data are common to both data sets.
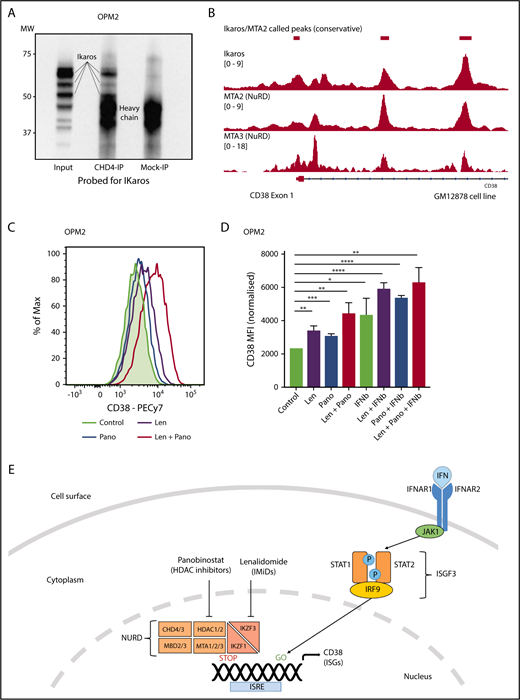
Ikaros represses CD38 through interaction with the NuRD complex. (A) Coimmunoprecipitation performed in OPM2 cells with anti-CHD4 (CHD4-IP) antibody or mouse-immunoglobulin G (Mock-IP) control. Membranes were probed for Ikaros. MW are shown on the left. Positions of the Ikaros and IgH proteins are indicated. (B) Analysis of Ikaros, MTA2, and MTA3 binding in the human B-cell line GM12878 by ChIP-seq from the ENCODE Project. Shown are the mapping track of sequence reads in each library at the CD38 locus. Ikaros conservatively called peaks are shown as bars in the top panel. (C) Histogram of CD38 expression by flow cytometry in OPM2 cells 48 hours after treatment with Len 1 μM and/or panobinostat (Pano) 10 nM or untreated control. (D) Graph of CD38 MFI in OPM2 cells 48 hours after treatment with Len 1 μM and/or Pano 10 nM and/or β-IFN (50 IU). Data are the MFI ± SD from 3 experiments. ****P < .0001, ***P < .001, **P < .01, *P < .05, using an unpaired Student t test. (E) Cartoon of model suggested: Transcriptional activation of ISGs, including CD38, results from signal transduction after the binding of interferon (α or β) to the type 1 interferon receptor, activation of JAK1, STAT1/2, and together with IRF9, assembly of the ISGF3 complex, which translocates to the nucleus and binds to IFN-stimulated response elements. Expression of ISGs is normally repressed to an extent by Ikaros and Aiolos, likely through interaction with the NuRD complex. Hence, CD38 expression can be augmented through treatment with IFN, IMiDs including lenalidomide and histone deacetylase inhibitors such as panobinostat.
Ikaros represses CD38 through interaction with the NuRD complex. (A) Coimmunoprecipitation performed in OPM2 cells with anti-CHD4 (CHD4-IP) antibody or mouse-immunoglobulin G (Mock-IP) control. Membranes were probed for Ikaros. MW are shown on the left. Positions of the Ikaros and IgH proteins are indicated. (B) Analysis of Ikaros, MTA2, and MTA3 binding in the human B-cell line GM12878 by ChIP-seq from the ENCODE Project. Shown are the mapping track of sequence reads in each library at the CD38 locus. Ikaros conservatively called peaks are shown as bars in the top panel. (C) Histogram of CD38 expression by flow cytometry in OPM2 cells 48 hours after treatment with Len 1 μM and/or panobinostat (Pano) 10 nM or untreated control. (D) Graph of CD38 MFI in OPM2 cells 48 hours after treatment with Len 1 μM and/or Pano 10 nM and/or β-IFN (50 IU). Data are the MFI ± SD from 3 experiments. ****P < .0001, ***P < .001, **P < .01, *P < .05, using an unpaired Student t test. (E) Cartoon of model suggested: Transcriptional activation of ISGs, including CD38, results from signal transduction after the binding of interferon (α or β) to the type 1 interferon receptor, activation of JAK1, STAT1/2, and together with IRF9, assembly of the ISGF3 complex, which translocates to the nucleus and binds to IFN-stimulated response elements. Expression of ISGs is normally repressed to an extent by Ikaros and Aiolos, likely through interaction with the NuRD complex. Hence, CD38 expression can be augmented through treatment with IFN, IMiDs including lenalidomide and histone deacetylase inhibitors such as panobinostat.
CD38 is the target of mAbs, including daratumumab, which has recently received approval in MM from the US Food and Drug Administration, as well as isatuximab, which was granted orphan drug status for this disease. Binding daratumumab to CD38 on MM cells leads to cell death predominantly through antibody-dependent cell-mediated cytotoxity (ADCC).39,43 Importantly, there is a direct correlation between the level of CD38 expression and drug efficacy in clinical studies.43 Although daratumumab has single-agent efficacy in relapsed-refractory disease, both preclinical and early clinical trials have suggested that outcomes are improved with combination treatment with lenalidomide.44-46 Thus far, this clinical synergy has been thought to be a result of the immune-enhancing action of the IMiDs on the effector cells mediating ADCC.47 In light of our observation of increased expression of CD38 after lenalidomide-mediated degradation of Ikaros and Aiolos, we formulated an alternate cell-intrinsic hypothesis that these treatments prime MM cells for daratumumab-induced ADCC. To test this hypothesis, H929 IK20 cells were treated with doxycycline (day 0, to inactivate Ikaros), lenalidomide (day 1 to degrade Ikaros/Aiolos protein), β-IFN (day 2, to upregulate CD38), or a combination of doxycycline + β-IFN or lenalidomide + β-IFN vs no treatment. At 96 hours, cells were washed, labeled, and treated with a dose titration of daratumumab before incubation with donor NK cells. Loss of Ikaros, or treatment with lenalidomide or β-IFN, each led to increased efficacy of ADCC (Figure 5E). Strikingly, an additive effect was seen with the combination treatments, directly correlating with the upregulation of CD38 surface expression (Figure 5C). These results were confirmed using a fixed dose of daratumumab and instead titrating the effector (NK cells) to target (H929) cell ratio (supplemental Figure 6D). Although daratumumab kills primarily through ADCC,39,43,48 additional reported mechanisms include complement-dependent cytotoxicity and direct cytotoxicity. We, however, did not observe any direct cytotoxicity in H929 or OPM2 cells across a broad dose titration of daratumumab, in the presence or absence of lenalidomide (supplemental Figure 7). Lenalidomide treatment of activated NK cells, which also express CD38,49 did not alter CD38 expression, despite a similar reduction in Ikaros, indicating that this process is specific to the MM cells (supplemental Figure 6E-F).
Ikaros represses CD38 through interaction with the NuRD complex
Ikaros has been widely documented to lead to transcriptional repression through interaction with the NuRD complex,50 although this association has not been demonstrated in MM cells. It was therefore of interest to us that a recent study demonstrated a similar increase in CD38 expression and subsequent daratumumab efficacy with the histone deacetylase inhibitor panobinostat.48 Given that HDAC1/2 are essential components of the NuRD complex,51,52 we hypothesized that Ikaros represses CD38 expression through interaction with this chromatin modifier. Coimmunoprecipitation using an antibody targeting CHD4 (a component of NuRD53 ) was able to pull down Ikaros in OPM2 cells, confirming the Ikaros/NuRD interaction in MM cells (Figure 6A). Furthermore, analysis of ChIPseq data in the GM12878 cells demonstrated markedly similar binding patterns for Ikaros, MTA2, and MTA3, 2 additional NuRD components, in the CD38 locus (Figure 6B). Finally, synergistic upregulation of CD38 was seen on combination treatment with lenalidomide and a low-dose panobinostat insufficient to lead to significantly increased expression as a single agent (Figure 6C-D). Collectively, these data support the conclusion that Ikaros directly represses CD38 expression through interaction with the NuRD complex (Figure 6E).
Discussion
The findings that the IMiDs lead to the cereblon-dependent degradation of Ikaros and Aiolos were important steps in improving our ability to understand and hence fully exploit the therapeutic potential of this class of drugs.1-4 However, several subsequent studies have reported additional mechanisms of action of the IMiDs in MM and other diseases, to some extent calling into question the central role of Ikaros and Aiolos degradation in their activity.15,16,54 Here we demonstrate that inactivation of either transcription factor alone recapitulates the cell intrinsic action of the IMiDs, resulting in cell cycle arrest and apoptosis. Furthermore, evaluation of the transcriptional changes resulting from their loss demonstrates striking overlap with lenalidomide treatment. Therefore, our data strongly support the central role of Ikaros and Aiolos loss in the action of the IMiDs. In contrast, our data do not support the model that the IRF4-MYC axis is the critical downstream target of Ikaros and Aiolos, and hence the IMiDs, in MM. Although this may be true in a subset of patients (and cell lines), and may depend on the genetic abnormalities within individual tumors, loss of MYC and IRF4 was not required for the cell death that ensued on deletion of Ikaros or Aiolos, and their overexpression failed to rescue the effects of Ikaros deficiency. Hence, Ikaros and Aiolos play critical roles in MM beyond the regulation of MYC and IRF4.
α-IFN has been demonstrated to have activity in MM, both upfront in combination with chemotherapy and as maintenance therapy.28-33 CC-122, an IMiD-like compound that also results in the cereblon-dependent degradation of Ikaros and Aiolos, was found to lead to the IRF4-independent upregulation of ISGs and cell death in DLBCL.26 Furthermore, upregulation of the IFN signaling pathway was identified by gene set enrichment analysis in a mouse xenograft MM model after IMiD and dexamethasone treatment, although the significance or mechanism of this finding was not explored.55 Our data suggest that this mechanism of action of the IMiDs extends to MM and that Ikaros and Aiolos potentially repress the expression of ISGs across a spectrum of B cell malignancies. Although it is tantalizing to hypothesize that combination treatment with lenalidomide (or CC-122) and IFN may result in synergistic activity in patients with MM (or DLBCL), as suggested by our in vitro data, the previous early trials of thalidomide combined with α-IFN were marred by severe toxicity and a lack of understanding on which patient subsets are likely to respond.56 Specific delivery of β-IFN to MM cells through oncolytic virus or conjugation of α-IFN to an anti-CD38 mAb has been shown to be effective in preclinical studies and may potentially avoid the toxicity of systemic administration.57,58
Our data confirm that CD38 is an ISG in the context of MM, and furthermore that its expression is repressed to an extent by Ikaros and Aiolos. Treatment with all trans retinoic acid was recently shown to lead to a similar upregulation of CD38 expression,41 indicating an overlap of function with IFN. Intriguingly, IFIT3, also known as retinoid acid induced gene G, is also upregulated on loss of Ikaros/Aiolos or treatment with lenalidomide. At this time, little is known of the overlap between the retinoic acid receptor and type I IFN signaling pathways or downstream stimulated genes. Similar to all trans retinoic acid treatment, the increased CD38 expression resulting from loss of Ikaros or treatment with lenalidomide or β-IFN primes MM cells for daratumumab-induced ADCC. These results provide a MM cell-intrinsic mechanism explaining the improved clinical results that have been reported for the treatment combination, lenalidomide and daratumumab.45 NK cells, known to express CD38, decline during daratumumab treatment. However, treatment efficacy is unaffected, likely because of the capacity of other immune cells to also mediate ADCC.49 Importantly, lenalidomide treatment did not result in increased CD38 expression on NK cells, and thus would not be expected to negatively affect host immune ADCC capacity. In fact, the clinical synergy seen with combination daratumumab and lenalidomide has been suggested to be secondary to the IMiD’s action on effector cells mediating ADCC,47 which may represent an additional benefit.
We have demonstrated that Ikaros interacts with CHD4, a component of the NuRD complex, in MM, and displays a remarkably similar binding pattern at the CD38 locus to that of MTA2/3, additional components of the NuRD complex. Given that HDAC1/2 are also integral NuRD components, our findings provide an explanation for the observation that treatment with the HDAC inhibitor panobinostat results in a similar upregulation of CD38 in MM and augmentation of daratumumab efficacy.48 As CD38 downregulation is likely to be an important mechanism in the development of disease resistance to anti-CD38 mAbs, it is clinically important to identify means to augment its expression. Collectively, our data provide preclinical evidence supporting combinational regimens including IMiDs and CD38 mAbs, as well as HDAC inhibitors and potentially IFN for the treatment of MM. In addition to the strong clinical data supporting combination treatment with daratumumab and lenalidomide,45 a recent study demonstrated efficacy with the combination panobinostat, lenalidomide, and weekly dexamethasone in relapsed refractory disease,59 with an objective response rate of 41%, including responses in patients with lenalidomide-refractory disease. Thus, both preclinical and clinical evidence support the investigation of the triplet combination of lenalidomide, panobinostat, and daratumumab in clinical trials, although toxicity would be predicted to be an obstacle limiting these approaches.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Lynn Corcoran and Stefan Glaser for reagents and Stephen Wilcox, Chris Riffkin, and Yuan Yao for technical assistance.
This project was supported by research grants from the National Health and Medical Research Council (NHMRC) Australia (1054618 and 1058238 to S.L.N, 1016701 to D.C.S.H.). The study was partly funded by the Cancer Council Victoria’s Grant-in-Aid Scheme (to S.L.N.). P.L.F. was supported by a Leukaemia Foundation of Australia Clinical Scholarship and a Royal College of Pathologists of Australasia Foundation Postgraduate Research Fellowship, S.N.W. by The Walter and Eliza Hall Trust Centenary Fellowship, M.S.L. by a CRB Blackburn scholarship (GNT1075151) jointly from the NHMRC Australia and Royal Australasian College of Physicians, W.S. was supported by a Walter and Eliza Hall Centenary Fellowship sponsored by Commonwealth Serum Laboratories Limited, and D.C.S.H. by an NHMRC Australia fellowship and funding from the Leukemia and Lymphoma Society Specialized Center of Research Grant (7001-13). This work was made possible through Victorian State Government Operational Infrastructure Support and NHMRC Independent Research Institute Infrastructure Support Scheme.
Authorship
Contribution: P.L.F. designed and performed virtually all experiments; S.N.W., M.S.L., J.R., D.H.S., and J.-N.G., assisted in some experiments and provided reagents; Y.L. and W.S. provided the bioinformatic analysis; N.D.H., D.C.S.H., G.G., J.T., and S.L.N. supervised the experimental design; and P.L.F. and S.L.N. wrote the manuscript with editorial input from all authors.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Pasquale L. Fedele, The Walter and Eliza Hall Institute, 1G Royal Parade, Parkville 3052, VIC, Australia; e-mail: fedele.p@wehi.edu.au; Stephen L. Nutt, The Walter and Eliza Hall Institute, 1G Royal Parade, Parkville 3052, VIC, Australia; e-mail: nutt@wehi.edu.au.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal