

In this issue of Blood, report that in chronic lymphocytic leukemia (CLL) patients, the γδ T cells are functionally compromised. These γδ T cells can recover their function and demonstrate cytotoxicity against tumor cells after in vitro activation/expansion and/or ibrutinib treatment, which can be further explored as immunotherapy for CLL patients.1
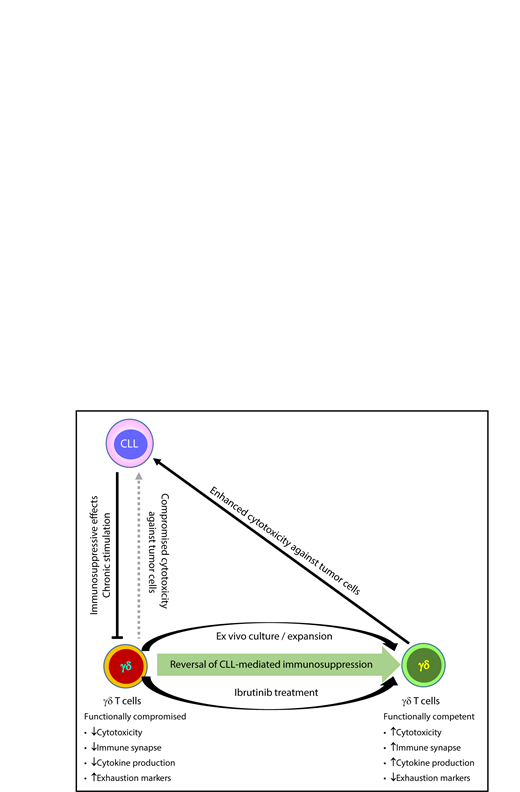
γδ T cells from CLL patients are functionally impaired and cannot efficiently lyse tumor cells. Their functional competency and cytotoxicity against CLL cells can be recovered after ex vivo stimulation or ibrutinib treatment.
γδ T cells from CLL patients are functionally impaired and cannot efficiently lyse tumor cells. Their functional competency and cytotoxicity against CLL cells can be recovered after ex vivo stimulation or ibrutinib treatment.
Most T cells are αβ T cells, which have T-cell receptors (TCRs) composed of α and β TCR chains. γδ T lymphocytes, on the other hand, have TCRs made up of γ and δ chains. Up to 10% of T cells in peripheral blood are γδ T cells. Unlike the conventional αβ T cells, γδ T cells have the unique capability to recognize and kill cells under stressed conditions, demonstrating both innate and adaptive immune properties.2 For example, under stressed conditions, γδ T cells recognize phosphoantigens that are produced as intermediate metabolites during cellular stress and malignant transformation. One major limitation for immunotherapy with conventional αβ T cells is that each clone of tumor-specific T cells can only recognize a single antigen epitope, in a HLA-restricted manor. For example, αβ T cells have been engineered to express TCRs specific for NY-ESO-1 or MART-1 (both are tumor-associated antigens). However, these αβ T cells can only recognize tumor cells that are of certain HLA types (eg, HLA-A2) and express high levels of the cognate antigens.3 γδ T cells, on the other hand, hunt and kill tumor cells in a manner similar to innate immune cells: they are not major histocompatibility complex restricted and are capable of recognizing a broader spectrum of tumor cells rather than a single specific antigen epitope.2 These features make γδ T cells a more “flexible” approach for cancer immunotherapy, especially for diseases like CLL, which is known to have limited options for tumor-associated antigens or mutation-specific “neo”-antigens and does not respond well to checkpoint blockade.4 In this paper, the authors reported the potential of ex vivo activated γδ T cells in immunotherapy of CLL patients.
CLL cells profoundly suppress T-cell immunity via multiple mechanisms, such as expression of inhibitory surface molecules (eg, CD200, HLA-G, and PD-L1), production of immunosuppressive cytokines, and induction of regulatory T cells and myeloid-derived suppressor cells.5 Dysfunction of “conventional” αβ T cells in CLL patients has been well studied. In CLL patients, these T cells demonstrate features of “exhaustion,” with significant upregulation of checkpoint molecules and exhaustion markers such as PD-1 and CD160. T-cell “exhaustion” usually results from repeated chronic stimulation.6 It has been postulated that CLL cells may cause chronic aberrant stimulation of T cells by inducing a CLL-specific immune response, or by modifying T-cell response to chronic infections such as cytomegalovirus.7 In line with this, CLL patients’ T-cell subsets are skewed toward a terminally differentiated phenotype, with significant reduction in naive T cells and expansion of more terminal differentiated effector memory/effector T cells. CLL also causes profound functional defects in T cells, including reduced cytotoxicity of CD8 T cells and defective immunologic synapse formation. T-cell response and functional status are skewed toward the Th2 polarization, which is considered to be detrimental in the scenario of antitumor immunity. However, little is known about how CLL affects γδ T cells.
In this paper, the authors reported that, in CLL patients, phenotypic and functional aberrations similar to those that occur in αβ T cells also occur in γδ T cells. They also demonstrated an exhausted phenotype, with subset distribution skewed toward a more terminal differentiated effector memory phenotype. Functionality wise, the γδ T cells have compromised cytokine production capability and limited cytotoxicity against CLL cells. Strikingly, the abovementioned dysfunctions can be induced in normal γδ T cells after being cocultured with CLL cells, which strongly suggests leukemia induced immune suppression. Moreover, γδ T-cell dysfunctions in CLL patients can be reversed after in vitro stimulation and both of the in vitro stimulation methods tested by the authors can also be used for clinical scale expansion for immunotherapy (see figure).
Ibrutinib, designed as a Bruton tyrosine kinase inhibitor for treatment of B-cell malignancies, has been found to have favorable immunomodulatory effects in multiple tumor models.8 For conventional αβ T cells, ibrutinib enhances Th1/Tc1 response9 in part through inhibition of interleukin-2 related tyrosine kinase (ITK). It also promotes the expansion and persistence of activated T cells by protecting them from activation-induced cell death.10 In this paper, the authors demonstrated similar findings for γδ T cells: ibrutinib binds to ITK in γδ T cells, and in vitro treatment with ibrutinib enhances markers of Th1/Tc1-like function and cytotoxicity in γδ T cells.
These new data suggest that γδ T cells could be a promising approach for immunotherapy in CLL patients; however, there are still questions to be addressed. Unlike αβ T cells, much less is known about γδ T cells. For example, the mechanism by which γδ T cells recognize targets is different. How costimulatory/coinhibitor signals regulate the activation vs tolerance of γδ T cells is also less understood. We cannot simply extrapolate the findings from αβ T cells to γδ T cells. For safe immunotherapies, it is crucial to ensure that the tumor cells are killed efficiently while the normal tissue/cells are spared (ie, a safe therapeutic window). More mechanistic studies are needed to understand how to test and improve γδ T cells’ specificity and efficacy against autologous tumor cells while sparing normal organs/cell types. In addition, for an immunotherapy to achieve clinical efficacy, the effector cells have to localize to the tumor site and battle with the immunosuppressive tumor microenvironment. Again, because most of our knowledge on these issues is from conventional αβ T cells, much more research about γδ T cells is needed.
Conflict-of-interest disclosure: The author declares no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal