

Key Points
Healthy Vγ9Vδ2-T cells recognize and lyse CLL cells, but CLL-derived Vγ9Vδ2-T cells have impaired cytotoxicity and cytokine production.
Vγ9Vδ2-T–cell dysfunction is reversible upon ex vivo activation with autologous moDCs, and ibrutinib promotes an antitumor TH1 phenotype.

Abstract
The efficacy of autologous (αβ) T-cell–based treatment strategies in chronic lymphocytic leukemia (CLL) has been modest. The Vγ9Vδ2-T cell subset consists of cytotoxic T lymphocytes with potent antilymphoma activity via a major histocompatibility complex–independent mechanism. We studied whether Vγ9Vδ2-T cells can be exploited as autologous effector lymphocytes in CLL. Healthy control Vγ9Vδ2-T cells were activated by and had potent cytolytic activity against CLL cells. However, CLL-derived Vγ9Vδ2-T cells proved dysfunctional with respect to effector cytokine production and degranulation, despite an increased frequency of the effector-type subset. Consequently, cytotoxicity against malignant B cells was hampered. A comparable dysfunctional phenotype was observed in healthy Vγ9Vδ2-T cells after coculture with CLL cells, indicating a leukemia-induced mechanism. Gene-expression profiling implicated alterations in synapse formation as a conceivable contributor to compromised Vγ9Vδ2-T–cell function in CLL patients. Dysfunction of Vγ9Vδ2-T cells was fully reversible upon activation with autologous monocyte-derived dendritic cells (moDCs). moDC activation resulted in efficient expansion and predominantly yielded Vγ9Vδ2-T cells with a memory phenotype. Furthermore, ibrutinib treatment promoted an antitumor T helper 1 (TH1) phenotype in Vγ9Vδ2-T cells, and we demonstrated binding of ibrutinib to IL-2-inducible kinase (ITK) in Vγ9Vδ2-T cells. Taken together, CLL-mediated dysfunction of autologous Vγ9Vδ2-T cells is fully reversible, resulting in potent cytotoxicity toward CLL cells. Our data support the potential use of Vγ9Vδ2-T cells as effector T cells in CLL immunotherapy and favor further exploration of combining Vγ9Vδ2-T-cell–based therapy with ibrutinib.
Introduction
Although novel drugs that inhibit key kinases of the B-cell receptor (BCR) signaling pathway are valuable additions to the therapeutic arsenal of chronic lymphocytic leukemia (CLL), these agents are not curative; continuous treatment is required, which invokes toxicities and resistance.1
Allogeneic hematopoietic stem cell therapy has demonstrated that T-cell–based therapy has curative potential in CLL.2,3 However, current autologous T-cell–based approaches, such as checkpoint inhibition and chimeric antigen receptor T cells, have yielded limited response rates in CLL,4-8 specifically when compared with their activity in more aggressive lymphoproliferative diseases (eg, acute lymphocytic leukemia).9-11 Activation of autologous T cells is constrained by low immunogenicity of CLL cells and by acquired T-cell dysfunction that progresses throughout the disease.12 T-cell abnormalities include altered cytokine-secretion profiles, an exhausted phenotype, and compromised cytotoxicity of CD8+ T cells,13,14 in addition to a subset distribution that is skewed toward an effector memory phenotype, particularly in cytomegalovirus (CMV)+ patients.15
A total of 1% to 10% of CD3+ T cells in the peripheral blood (PB) carries a highly conserved γδ T-cell receptor (TCR). Vγ9Vδ2-T cells form the predominant γδ T-cell subset in PB. In contrast to αβ T-cell antigen recognition, Vγ9Vδ2-T cells respond to stress molecules in malignant cells, in a TCR-dependent, yet major histocompatibility complex–independent, process.16,17 The Vγ9Vδ2-TCR is activated via nonpeptidic phosphorylated antigens, or phosphoantigens, produced as intermediate metabolites in the mevalonate pathway during cellular stress, malignant transformation, or upon treatment with aminobisphosphonates.18,19 Upon activation, Vγ9Vδ2-T cells have potent cytolytic activity against a wide variety of malignant cells, including lymphoma cells.20-24 Moreover, strategies based on Vγ9Vδ2-T cells, rather than total CD3 activation, have low toxicity.24-30 Therefore, Vγ9Vδ2-T cells could represent an attractive alternative source of autologous effector T cells for CLL immunotherapy.20-24
In support of the relevance of Vγ9Vδ2-T cells in CLL, Coscia et al have previously described a correlation between a low proliferative response of Vγ9Vδ2-T cells to the aminobisphosphonate zolendronate and short time to first treatment status in CLL.31 Diminished proliferation was mostly seen in CLL patients with unmutated immunoglobulin genes (U-CLL), who had higher phosphoantigen levels and a hyperactive mevalonate pathway in comparison with patients with CLL with mutated immunoglobulin genes. The mechanism responsible for the shorter time to first treatment and the ability of Vγ9Vδ2-T cells from CLL patients to eradicate leukemic cells remain unexplored. Moreover, little is known about functional characteristics of this subset beyond proliferation and whether perturbations demonstrated in αβ T cells extend to Vγ9Vδ2-T cells.
Therefore, we extensively characterized CLL-derived Vγ9Vδ2-T cells and studied whether they can be exploited as effector lymphocytes in CLL.
Materials and methods
Patient material
PB mononuclear cells (PBMCs) were isolated from PB samples from untreated CLL patients or from age-matched healthy controls (HCs) or from HC buffy coats from Sanquin Blood Supply (Amsterdam, The Netherlands) and cryopreserved as described previously (Table 1).32 The presence of monoclonal B-cell lymphocytosis was excluded in HCs by CD5, CD19, κ, and λ immunophenotyping. The study was approved by the medical ethics committee at the Academic Medical Center. Written informed consent was obtained from all subjects in accordance with the Declaration of Helsinki.
Patient characteristics
| Immunophenotype | Cytokine assay | Cytotoxicity assay | Ex vivo activation | |
|---|---|---|---|---|
| HCs | ||||
| No. | 20 | 12 | 5 | 4 |
| Males, % | 45 | 58 | 60 | 50 |
| Age, mean (range), y | 72 (53-84) | 67 (53-79) | 68 (62-73) | 67 (62-73) |
| Vγ9Vδ2, mean (range), % of CD3 | 1.4 (0.1-4.9) | 1.5 (0.1-4.9) | 1.2 (0.5-1.5) | 1.2 (0.5-1.5) |
| CMV+, % | 55 | 50 | 40 | 50 |
| CLL patients | ||||
| No. | 39 | 14 | 5 | 8 |
| Males, % | 67 | 50 | 60 | 50 |
| Age, mean (range), y | 64 (41-87) | 67 (55-83) | 64 (57-69) | 64.9 (57-69) |
| Vγ9Vδ2, mean (range), % of CD3 | 1.0 (0.1-6.2) | 0.8 (0.1-1.8) | 1.3 (0.6-2.6) | 1.2 (0.6-2.6) |
| CMV+, % | 64.3 | 50 | 60 | 62.5 |
| ALC, mean (range), ×109 cells/L | 80.4 (15.3-358.7) | 77.4 (19.8-226.1) | 51.3 (28.0-95.6) | 44.3 (10.6-95.6) |
| U-CLL, % | 43.6 | 42.9 | 40 | 50 |
| Rai stage 0+1, % | 72 | 64 | 60 | 62.5 |
| Immunophenotype | Cytokine assay | Cytotoxicity assay | Ex vivo activation | |
|---|---|---|---|---|
| HCs | ||||
| No. | 20 | 12 | 5 | 4 |
| Males, % | 45 | 58 | 60 | 50 |
| Age, mean (range), y | 72 (53-84) | 67 (53-79) | 68 (62-73) | 67 (62-73) |
| Vγ9Vδ2, mean (range), % of CD3 | 1.4 (0.1-4.9) | 1.5 (0.1-4.9) | 1.2 (0.5-1.5) | 1.2 (0.5-1.5) |
| CMV+, % | 55 | 50 | 40 | 50 |
| CLL patients | ||||
| No. | 39 | 14 | 5 | 8 |
| Males, % | 67 | 50 | 60 | 50 |
| Age, mean (range), y | 64 (41-87) | 67 (55-83) | 64 (57-69) | 64.9 (57-69) |
| Vγ9Vδ2, mean (range), % of CD3 | 1.0 (0.1-6.2) | 0.8 (0.1-1.8) | 1.3 (0.6-2.6) | 1.2 (0.6-2.6) |
| CMV+, % | 64.3 | 50 | 60 | 62.5 |
| ALC, mean (range), ×109 cells/L | 80.4 (15.3-358.7) | 77.4 (19.8-226.1) | 51.3 (28.0-95.6) | 44.3 (10.6-95.6) |
| U-CLL, % | 43.6 | 42.9 | 40 | 50 |
| Rai stage 0+1, % | 72 | 64 | 60 | 62.5 |
ALC, absolute leukocyte count.
Flow cytometry
Thawed PBMCs were stained with monoclonal antibodies (details can be found in supplemental Methods and supplemental Table 1, available on the Blood Web site) and measured on an LSR Fortessa cytometer (BD Biosciences). Samples were analyzed with FlowJo for Mac v10.
Cytotoxicity
CD3+TCR-Vγ9+TCR-Vδ2+ cells were sorted from PBMCs and depleted of CD19+ cells with magnetic beads (Miltenyi Biotec, Bergisch Gladbach, Germany; CD19 fraction <5%) on a FACSAria IIu 3-laser (BD Biosciences). Thawed allogeneic CLL PBMCs (>95% CD5+CD19+) or Daudi cells (American Type Culture Collection) were used as target cells; they were labeled with carboxyfluorescein succinimidyl ester (Thermo Fisher Scientific, Waltham, MA) and subsequently treated with 25 μM aminobisphosphonates (ABPs) for 2 hours (pamidronate; TEVA Pharmachemie, Haarlem, The Netherlands) as indicated before coculture with sorted Vγ9Vδ2-T cells (purity > 95%). Where indicated, Vγ9Vδ2-T cells were treated with 100 nM concanamycin A (Sigma-Aldrich, St. Louis, MO) or dimethyl sulfoxide control for 2 hours and washed before coculture.
Viability was measured using MitoTracker Orange (Invitrogen) and TO-PRO-3 (Invitrogen) on a FACSCanto flow cytometer system (BD Biosciences).
Cytokine and degranulation assays
Target cells, thawed allogeneic CLL PBMCs (CD5+CD19+ > 95%) or Daudi cells, were pretreated with 25 μM ABPs or medium for 2 hours. CD19-depleted PBMCs were stimulated with target cells at a 1:5 Vγ9Vδ2-T–cell effector-to-target cell ratio with anti-CD28 (2 µg/mL, 15E8; Sanquin) for 16 to 18 hours or with phorbol-12-myristate-13-acetate (PMA; 10 ng/mL; Sigma-Aldrich) and ionomycin (1 µg/mL; Sigma-Aldrich) for 4 hours at 37°C. Brefeldin A (10 µg/mL; Invitrogen), GolgiStop, and anti-CD107a allophycocyanin (BD Biosciences) were present during the final 4 to 6 hours.33,34
Where indicated, CD19-depleted PBMCs were treated with ibrutinib (Pharmacyclics, Sunnyvale, CA) or CC-292 (Selleckchem, Houston, TX) for 30 minutes before coculture.
Coculture assay
CLL or healthy B cells were isolated with anti-CD19 magnetic MicroBeads and cocultured with allogeneic CD19-depleted HC PBMCs for 36 hours in a 1:10 ratio. CD25 expression was measured, and cells were cocultured with ABP-pretreated Daudi cells or stimulated with PMA/ionomycin for cytokine and degranulation assays, as above.
moDC-based ex vivo Vγ9Vδ2-T–cell activation and expansion
CD3+TCR-Vγ9+TCR-Vδ2+ cells were sorted using fluorescence-activated cell sorting (FACS), as above, and cultured with monocyte-derived dendritic cells (moDCs), as previously described.35 In short, CD14+ cells were isolated from HC or CLL PBMCs (anti-CD14 magnetic MicroBeads; Miltenyi Biotec) and cultured for 7 days in the presence of IL-4 (20 ng/mL; R&D Systems, Minneapolis, MN) and granulocyte-macrophage colony-stimulating factor (100 U/mL; Genzyme, Cambridge, MA) (0.45 × 106 cells per milliliter) to generate immature moDCs. These immature moDCs were matured with lipopolysaccharide (100 ng/mL; Sigma-Aldrich) for 48 hours and ABPs (100 μM) during the final 2 hours. Mature moDCs were then irradiated (5000 rad) and cocultured with sorted Vγ9Vδ2-T cells in the presence of IL-7 (10 U/mL) and IL-15 (10 ng/mL, R&D Systems) for 2 weeks (1 × 106 Vγ9Vδ2-T cells and 0.2 × 106 moDCs per milliliter). Vγ9Vδ2-T cells were restimulated with irradiated moDCs weekly. When using autologous moDCs, cultures were performed in medium supplemented with 10% autologous filter-sterilized serum.
Vγ9Vδ2-T–cell generation from ABP/IL-2–treated PBMCs
CD19-depleted PBMCs were treated with ABPs (25 μM) and IL-2 (1000 IU/mL; PeproTech, Rocky Hill, NJ) for 2 weeks. The medium was refreshed with ABPs and IL-2–containing medium 3 times per week.
RNA sequencing and data analysis
RNA was isolated (NucleoSpin RNA kit; Macherey Nagel, Düren, Germany) from FACS-sorted CD3+TCR-Vγ9+TCR-Vδ2+ cells. Complementary DNA synthesis and amplification were performed with the Ovation RNA-Seq System V2, followed by library preparation with the Ovation Ultralow System V2 (both from Nugen, San Carlos, CA). Single-end 75–base pair sequencing was performed on an Illumina NextSeq 500 sequencer by GenomeScan (Leiden, The Netherlands).
RNA sequencing analyses were performed using R (v.3.4.3) and Bioconductor (v3.6) (see supplemental Methods). Gene set enrichment analysis was performed using CAMERA, with a combination of all Hallmark (collection H) and BioCarta (collection C2) gene sets retrieved from the Molecular Signatures Database (MSigDB v6.1; Entrez Gene ID version) and 10 manually generated (supplemental Table 2) gene sets. Sequence data have been deposited in the European Genome-phenome Archive under accession number EGAS00001003193.
Ibrutinib pull-down
Biotinylated ibrutinib derivative (1 µM)36 was coupled to avidin agarose (Thermo Fisher Scientific; 30 minutes at room temperature). Remaining binding sites were blocked with 10 mM biotin (Sigma-Aldrich; 15 minutes at room temperature). HC Vγ9Vδ2-T cells or Mec-1 (DSMZ) cells were lysed with NP-40 lysis buffer. Lysates were treated with 1 µM ibrutinib or 1 µM CC-292 (30 minutes at room temperature) and incubated overnight with ibrutinib-coupled or uncoupled control agarose at 4°C. Proteins were eluted by heating to 95°C in Laemmli buffer (60 mM Tris-HCl pH 6.8, 10% glycerol, 2% sodium dodecyl sulfate, 100 mM dithiothreitol) for 10 minutes and analyzed by western blotting, as described previously.37 Blots were probed with anti-human Bruton tyrosine kinase (BTK) (611116, BD Biosciences) or IL-2–inducible T-cell kinase (ITK; 2380S; Cell Signaling Technology, Danvers, MA).
Statistical analysis
Data were checked for normality with the D’Agostino-Pearson normality test and analyzed using 2-sided paired or unpaired t tests, the Mann-Whitney U test, or 1-way analysis of variance (ANOVA; followed by the Bonferroni or Dunnett post hoc test) as indicated, with significance set at P < .05, using GraphPad Prism 5. Statistical analyses of RNA sequencing data were performed using edgeR and limma R/Bioconductor packages (supplemental Methods). Data are presented as mean and standard error of the mean (SEM).
Results
Vγ9Vδ2-T cells are cytotoxic against CLL cells, yet cytotoxic function is impaired in CLL-derived Vγ9Vδ2-T cells
The capacity of CLL cells to activate Vγ9Vδ2-T cells was determined by measuring the expression of CD25 after coculture. HC-derived Vγ9Vδ2-T cells were activated by CLL cells, as demonstrated by robust induction of CD25 expression. In contrast, culture with allogeneic healthy B cells did not alter CD25 expression (Figure 1A-B).
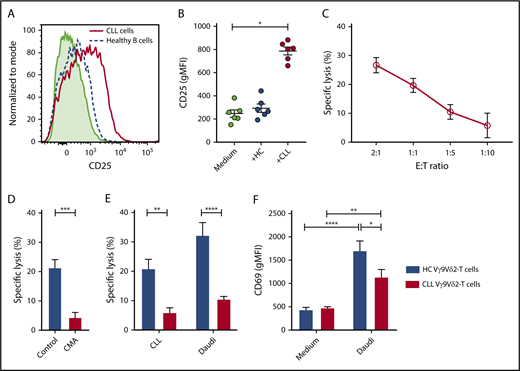
Vγ9Vδ2-T–cell recognition and lysis of CLL cells. (A) Representative plot of geometric mean fluorescence intensity (gMFI) of CD25 expression measured after a 36-hour coculture of HC Vγ9Vδ2-T cells with CLL cells (solid red line), allogeneic healthy B cells (dashed blue line), or Vγ9Vδ2-T cells alone (light green shading). (B) As in (A), scatter plot summarizing results for 6 donors. (C-E) Cell death of carboxyfluorescein succinimidyl ester–labeled target cells after overnight coculture with Vγ9Vδ2-T cells, measured by MitoTracker Orange and TO-PRO-3. (C) Specific lysis of CLL cells after coculture with Vγ9Vδ2-T cells from HCs at the indicated effector-to-target ratios (n = 9). Specific lysis was calculated as (% cell death in stimulated cells) − % cell death in unstimulated cells)/(% viable cells in unstimulated cells) * 100. (D) Healthy Vγ9Vδ2-T cells were treated for 2 hours with 100 nM concanamycin A (CMA) or dimethyl sulfoxide and washed before coculture with CLL cells at a 1:1 ratio (n = 9). (E) Specific lysis of allogeneic CLL and Daudi cells after coculture with Vγ9Vδ2-T cells from CLL patients (n = 6) or HCs (n = 9) at a 1:1 ratio. (F) CD69 expression on Vγ9Vδ2-T cells from CLL patients (n = 8) or HCs (n = 4) after overnight coculture with Daudi cells at a 1:5 ratio. Data are mean and SEM. *P < .05, **P < .01, ***P < .001, ****P < .0001, 1-way ANOVA, followed by the Dunnett (B) or Bonferroni (E-F) correction; paired t test (D).
Vγ9Vδ2-T–cell recognition and lysis of CLL cells. (A) Representative plot of geometric mean fluorescence intensity (gMFI) of CD25 expression measured after a 36-hour coculture of HC Vγ9Vδ2-T cells with CLL cells (solid red line), allogeneic healthy B cells (dashed blue line), or Vγ9Vδ2-T cells alone (light green shading). (B) As in (A), scatter plot summarizing results for 6 donors. (C-E) Cell death of carboxyfluorescein succinimidyl ester–labeled target cells after overnight coculture with Vγ9Vδ2-T cells, measured by MitoTracker Orange and TO-PRO-3. (C) Specific lysis of CLL cells after coculture with Vγ9Vδ2-T cells from HCs at the indicated effector-to-target ratios (n = 9). Specific lysis was calculated as (% cell death in stimulated cells) − % cell death in unstimulated cells)/(% viable cells in unstimulated cells) * 100. (D) Healthy Vγ9Vδ2-T cells were treated for 2 hours with 100 nM concanamycin A (CMA) or dimethyl sulfoxide and washed before coculture with CLL cells at a 1:1 ratio (n = 9). (E) Specific lysis of allogeneic CLL and Daudi cells after coculture with Vγ9Vδ2-T cells from CLL patients (n = 6) or HCs (n = 9) at a 1:1 ratio. (F) CD69 expression on Vγ9Vδ2-T cells from CLL patients (n = 8) or HCs (n = 4) after overnight coculture with Daudi cells at a 1:5 ratio. Data are mean and SEM. *P < .05, **P < .01, ***P < .001, ****P < .0001, 1-way ANOVA, followed by the Dunnett (B) or Bonferroni (E-F) correction; paired t test (D).
The cytotoxic potency of Vγ9Vδ2-T cells against CLL was studied by measuring target cell death after coculture of sorted HC Vγ9Vδ2-T cells with CLL cells. The Burkitt lymphoma cell line Daudi was used as a positive control target (supplemental Figure 1A). Vγ9Vδ2-T cells from HCs induced CLL cell death (Figure 1C). Cell death was granzyme dependent because pretreatment of Vγ9Vδ2-T cells with concanamycin A, which prevents granule exocytosis, abrogated target cell death (Figure 1D).
Similarly, the cytotoxicity of CLL-derived Vγ9Vδ2-T cells toward allogeneic CLL cells was measured. Vγ9Vδ2-T cells from CLL patients were significantly less effective at inducing cell death in CLL and Daudi target cells (Figure 1E).
To assess whether the impaired cytotoxicity of CLL-derived Vγ9Vδ2-T cells reflects diminished activation, expression of the activation marker CD69 was measured after coculture with Daudi cells. V9Vδ2-T cells from CLL patients were activated by the Daudi cells but not as strongly as were the HC Vγ9Vδ2-T cells (Figure 1F).
Taken together, these results indicate activation of Vγ9Vδ2-T cells by CLL, resulting in a granzyme-dependent cytotoxic response. However, cytotoxic function is impaired in Vγ9Vδ2-T cells from CLL patients, suggesting phenotypic and functional alterations of Vγ9Vδ2-T cells in the context of CLL.
CLL-derived Vγ9Vδ2-T cells are more differentiated and express less granzyme B
A phenotypical analysis of CLL-derived vs HC-Vγ9Vδ2-T cells was performed directly ex vivo. The absolute number as well as the proportion of Vγ9Vδ2-T cells was comparable in CLL patients and age-matched HCs (Figure 2A-B). Vγ9Vδ2-T cells can be divided into naive, central memory, effector memory (EM), and RA re-expressing effector memory (EMRA) subsets based on the surface expression of CD27 and CD45RA.38-40 Functionally, central memory and naive cells have a larger proliferative response, whereas EM and EMRA cells have a higher cytokine production and cytotoxic capacity. CLL-derived Vγ9Vδ2-T–cell subsets were skewed, with a higher prevalence of EM cells, at the expense of naive-type cells (Figure 2C; supplemental Figure 1B-C). In CD8+ T cells from CLL patients, subset distribution is skewed, particularly in CMV+ patients.15 The subset distribution of Vγ9Vδ2-T cells was similar in CMV+ and CMV− CLL patients (supplemental Figure 1D-E).
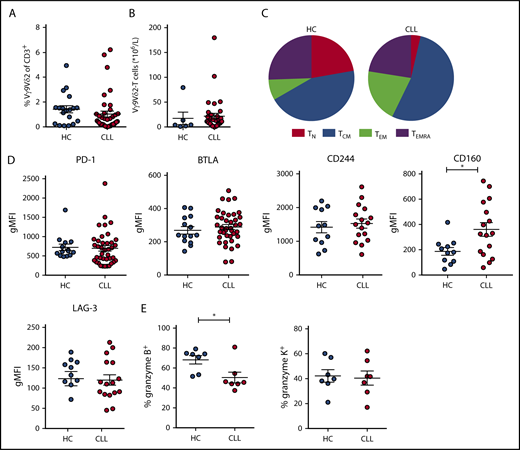
Frequency and immune phenotype of Vγ9Vδ2-T cells in CLL patients and HCs. Immunophenotyping of Vγ9Vδ2-T cells from untreated CLL patients and age-matched HCs, directly ex vivo. (A) Frequency of Vγ9+Vδ2+ cells within CD3+ T lymphocytes (CLL, n = 39; HC, n = 20). (B) Absolute number of Vγ9Vδ2-T cells (CLL, n = 39; HC, n = 6). (C) Distribution of differentiation subsets within Vγ9Vδ2-T cells based on CD27 and CD45RA expression (CLL, n = 39; HC, n = 20). (D) Expression of exhaustion markers on Vγ9Vδ2-T cells. Geometric mean fluorescence intensity (gMFI) of PD-1 and BTLA expression (CLL, n = 39; HC, n = 20) and CD244, CD160, and LAG-3 expression (CLL, n = 16; HC, n = 11). (E) Frequency of granzyme B+ and granzyme K+ cells within Vγ9Vδ2-T cells (CLL, n = 7; HC, n = 7). Data are mean and SEM. *P < .05, Student t test. TCM, CD27+CD45RA−; TEM, CD27−CD45RA−; TEMRA, CD27−CD45RA+; TN, CD27+CD45RA+.
Frequency and immune phenotype of Vγ9Vδ2-T cells in CLL patients and HCs. Immunophenotyping of Vγ9Vδ2-T cells from untreated CLL patients and age-matched HCs, directly ex vivo. (A) Frequency of Vγ9+Vδ2+ cells within CD3+ T lymphocytes (CLL, n = 39; HC, n = 20). (B) Absolute number of Vγ9Vδ2-T cells (CLL, n = 39; HC, n = 6). (C) Distribution of differentiation subsets within Vγ9Vδ2-T cells based on CD27 and CD45RA expression (CLL, n = 39; HC, n = 20). (D) Expression of exhaustion markers on Vγ9Vδ2-T cells. Geometric mean fluorescence intensity (gMFI) of PD-1 and BTLA expression (CLL, n = 39; HC, n = 20) and CD244, CD160, and LAG-3 expression (CLL, n = 16; HC, n = 11). (E) Frequency of granzyme B+ and granzyme K+ cells within Vγ9Vδ2-T cells (CLL, n = 7; HC, n = 7). Data are mean and SEM. *P < .05, Student t test. TCM, CD27+CD45RA−; TEM, CD27−CD45RA−; TEMRA, CD27−CD45RA+; TN, CD27+CD45RA+.
Similar to αβ T cells, the expression of exhaustion markers correlates with dysfunction in Vγ9Vδ2-T cells.41,42 Therefore, expression of PD-1, BTLA, CD160, CD244, and LAG-3 was determined (Figure 2D). Of these markers, only CD160 expression was higher on CLL Vγ9Vδ2-T cells compared with HC Vγ9Vδ2-T cells. There were no significant differences between CMV+ and CMV− patients with regard to the expression of exhaustion markers (supplemental Figure 1D-E).
To obtain mechanistic insight into the impaired cytotoxic function of CLL-derived Vγ9Vδ2-T cells, the cytotoxic effector molecules granzyme B and K were quantified in unstimulated Vγ9Vδ2-T cells. CLL-derived Vγ9Vδ2-T cells contained less granzyme B than HC-derived Vγ9Vδ2-T cells. In contrast, the percentage of Vγ9Vδ2-T cells expressing granzyme K did not differ between HCs and CLL patients (Figure 2E).
The function of Vγ9Vδ2-T cells can be attenuated through natural killer cell receptors; in particular, NKG2D promotes effector functions.43 The functional impairments observed in Vγ9Vδ2-T cells from CLL patients do not merely reflect lower NKG2D expression levels, which were, in fact, somewhat higher on CLL Vγ9Vδ2-T cells (supplemental Figure 1G).
Diminished effector-type cytokine production and cytotoxic potential of CLL-derived Vγ9Vδ2-T cells
Next, functional differences in cytokine production between HC and CLL Vγ9Vδ2-T cells were determined. Upon TCR-independent stimulation with PMA/ionomycin, Vγ9Vδ2-T cells from HCs and CLL patients produced mainly effector-type cytokines (IFN-γ and TNF-α) (Figure 3A). Significantly fewer CLL Vγ9Vδ2-T cells produced IFN-γ (30.0% vs 51.4%) and TNF-α (30.0% vs 55.2%) compared with their healthy counterparts. Because IFN-γ and TNF-α production was diminished, the possibility of T helper 2 (TH2) skewing was analyzed. Vγ9Vδ2-T cells from CLL patients produced as much IL-4 as HCs (Figure 3A).
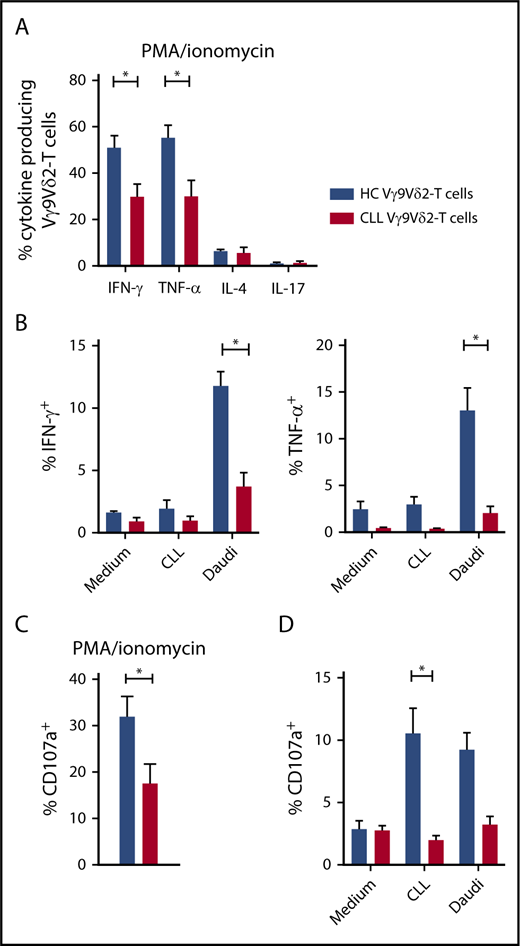
Impaired production of effector cytokines and degranulation in CLL-derived Vγ9Vδ2-T cells. (A) Production of IFN-γ, TNF-α, IL-17 (CLL, n = 14; HC, n = 12), and IL-4 (CLL and HC, n = 4) by Vγ9Vδ2-T cells after stimulation with PMA/ionomycin for 4 hours. (B) CD19-depleted PBMCs from CLL patients (n = 8) or HCs (n = 8) were cocultured with allogeneic CLL cells or Daudi cells for 16 to 18 hours. During the last 6 hours of coculture, brefeldin A and GolgiStop were added to measure cytokine production in Vγ9Vδ2-T cells. (C) CD107a expression in Vγ9Vδ2-T cells after PMA/ionomycin stimulation for 4 hours (CLL, n = 14; HC, n = 12). (D) CD107a expression after coculture with malignant B cells as in (B). Data are mean and SEM. *P < .05, 1-way ANOVA, followed by the Bonferroni post hoc test (A), Student t test (B-D).
Impaired production of effector cytokines and degranulation in CLL-derived Vγ9Vδ2-T cells. (A) Production of IFN-γ, TNF-α, IL-17 (CLL, n = 14; HC, n = 12), and IL-4 (CLL and HC, n = 4) by Vγ9Vδ2-T cells after stimulation with PMA/ionomycin for 4 hours. (B) CD19-depleted PBMCs from CLL patients (n = 8) or HCs (n = 8) were cocultured with allogeneic CLL cells or Daudi cells for 16 to 18 hours. During the last 6 hours of coculture, brefeldin A and GolgiStop were added to measure cytokine production in Vγ9Vδ2-T cells. (C) CD107a expression in Vγ9Vδ2-T cells after PMA/ionomycin stimulation for 4 hours (CLL, n = 14; HC, n = 12). (D) CD107a expression after coculture with malignant B cells as in (B). Data are mean and SEM. *P < .05, 1-way ANOVA, followed by the Bonferroni post hoc test (A), Student t test (B-D).
Recently, a tumor-promoting role has been attributed to IL-17–producing γδ T cells in solid malignancies.44 The percentage of IL-17–producing Vγ9Vδ2-T cells was negligible in HCs and CLL patients following PMA/ionomycin stimulation (Figure 3A).
Cytokine production upon contact with malignant B cells was then measured in Vγ9Vδ2-T cells. Coculture with Daudi cells triggered production of IFN-γ and TNF-α (Figure 3B). IFN-γ and TNF-α production was significantly impaired in CLL Vγ9Vδ2-T cells compared with HC Vγ9Vδ2-T cells.
Moreover, expression of CD107a, a marker for degranulation, was significantly lower in Vγ9Vδ2-T cells from CLL patients in response to PMA/ionomycin (Figure 3C), as well as CLL cells (Figure 3D).
In summary, the production of IFN-γ and TNF-α, as well as degranulation in response to TCR-independent stimulation and malignant B cells, is diminished in CLL Vγ9Vδ2-T cells, resulting in a diminished TH1 response and cytotoxic capability.
CLL cells can induce dysfunction in healthy Vγ9Vδ2-T cells
We hypothesized that functional impairments in Vγ9Vδ2-T cells result from CLL-related immune suppression rather than an intrinsic Vγ9Vδ2-T–cell defect. This was supported by the observation that cytotoxicity (supplemental Figure 2A), effector-type cytokine production (supplemental Figure 2B), and degranulation (supplemental Figure 2C) correlated inversely with absolute leukocyte count. The cytotoxic capacity, degranulation, and effector-type cytokine production of Vγ9Vδ2-T cells from U-CLL patients and patients with CLL with mutated immunoglobulin genes was comparable (supplemental Figure 2D-F).
To test whether CLL cells could induce functional impairments in healthy Vγ9Vδ2-T cells, HC Vγ9Vδ2-T cells were cultured for 48 hours in the presence of CLL cells or allogeneic healthy B cells. After subsequent coculture with Daudi cells as targets, Vγ9Vδ2-T cells that were cocultured with CLL cells produced significantly less IFN-γ and TNF-α than did Vγ9Vδ2-T cells that were cocultured with healthy B cells or medium alone (Figure 4A). Similarly, degranulation was impaired in CLL-cocultured Vγ9Vδ2-T cells (Figure 4B).
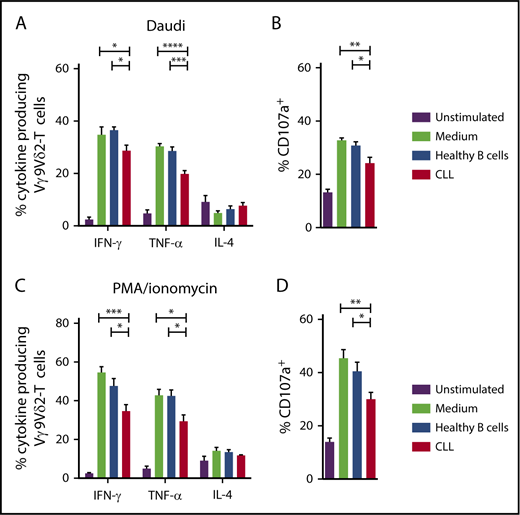
CLL cells induce Vγ9Vδ2-T–cell dysfunction. Cytokine production (A) and CD107a expression (B) by HC Vγ9Vδ2-T cells after coculture with allogeneic healthy B cells or CLL cells for 36 hours at a 1:10 ratio and subsequent coculture with ABP-pretreated Daudi cells (n = 6). Cytokine production (C) and CD107a expression (D) by HC Vγ9Vδ2-T cells after coculture and subsequent stimulation with PMA/ionomycin (n = 6). Data are mean and SEM. *P < .05, **P < .01, ***P < .001, ****P < .0001, 1-way ANOVA, followed by the Bonferroni post hoc test.
CLL cells induce Vγ9Vδ2-T–cell dysfunction. Cytokine production (A) and CD107a expression (B) by HC Vγ9Vδ2-T cells after coculture with allogeneic healthy B cells or CLL cells for 36 hours at a 1:10 ratio and subsequent coculture with ABP-pretreated Daudi cells (n = 6). Cytokine production (C) and CD107a expression (D) by HC Vγ9Vδ2-T cells after coculture and subsequent stimulation with PMA/ionomycin (n = 6). Data are mean and SEM. *P < .05, **P < .01, ***P < .001, ****P < .0001, 1-way ANOVA, followed by the Bonferroni post hoc test.
Production of effector-type cytokines (Figure 4C) and degranulation (Figure 4D) were also impaired upon TCR-independent stimulation with PMA/ionomycin in healthy Vγ9Vδ2-T cells that were cocultured with CLL cells. IL-4 production was not affected by CLL coculture.
In conclusion, CLL cells can induce dysfunction in healthy Vγ9Vδ2-T cells.
ABP pretreatment of CLL cells modestly augments cytotoxicity
Because ABP treatment can increase target cell recognition by Vγ9Vδ2-T cells through upregulation of phosphoantigen levels,18 and ABPs were previously shown to increase phosphoantigen production by CLL cells,31 we examined whether ABP treatment of target cells could overcome the observed dysfunction in CLL-derived Vγ9Vδ2-T cells.
First, we confirmed the expression of CD277/BTN3A1, the transmembrane protein required for phosphoantigen recognition,18 on CLL cells (supplemental Figure 3A-B). Pretreatment of Daudi cells with ABPs led to more IFN-γ–producing Vγ9Vδ2-T cells and lysis of Daudi cells (supplemental Figure 3C-F). In contrast, ABP pretreatment of CLL cells generated only a trend toward increased IFN-γ and TNF-α production by CLL-derived Vγ9Vδ2-T cells (supplemental Figure 3C-D), yet it did enhance degranulation (supplemental Figure 3E). Correspondingly, CLL-derived Vγ9Vδ2-T cells lysed allogeneic CLL cells more efficiently after ABP treatment (supplemental Figure 3F), although not to the level of HC Vγ9Vδ2-T cells (Figure 1E).
Thus, ABP pretreatment of CLL cells has a modest sensitizing effect on lysis by Vγ9Vδ2-T cells in the context of dysfunctional Vγ9Vδ2-T cells from CLL patients.
Vγ9Vδ2-T–cell dysfunction is reversible upon ex vivo activation
To examine the plasticity of Vγ9Vδ2-T–cell dysfunction and the feasibility of Vγ9Vδ2-T–cell expansion in CLL patients, sorted Vγ9Vδ2-T cells from HCs or CLL patients were activated ex vivo by culturing them for 2 weeks with phosphoantigen-expressing HC-derived moDCs in the presence of IL-7 and IL-15.35 Samples from patients with a previously confirmed dysfunctional phenotype (Figures 1E and 3) were used.
Following ex vivo culture, cytokine production and degranulation upon coculture with Daudi cells were measured. The percentage of Vγ9Vδ2-T cells that produced IFN-γ, TNF-α, or IL-4 did not differ between CLL- and HC-derived cultured Vγ9Vδ2-T cells (supplemental Figure 4A), which was also true with regard to degranulation (supplemental Figure 4B). Furthermore, cultured Vγ9Vδ2-T cells from HCs and CLL patients killed an equal amount of Daudi cells (supplemental Figure 4C). Thus, Vγ9Vδ2-T–cell dysfunction in CLL patients is fully reversible upon ex vivo activation.
To adapt this activation method to a clinically achievable setting, the reversibility of the dysfunction was subsequently tested using autologous moDCs and serum. The proliferative capacity of Vγ9Vδ2-T cells was similar with allogeneic and autologous moDCs (mean expansion factor 87.00 ± 16.46 vs 76.42 ± 8.55) and was not impaired in CLL-derived Vγ9Vδ2-T cells in comparison with HC-derived Vγ9Vδ2-T cells (Figure 5A). Vγ9Vδ2-T cells from CLL patients and HCs were equally activated after culture, as measured by CD25 expression (supplemental Figure 4D).
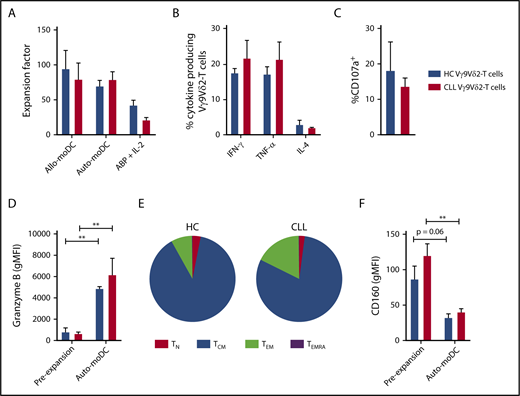
Vγ9Vδ2-T–cell dysfunction is reversible upon ex vivo activation and expansion. Vγ9Vδ2-T cells from CLL patients (n = 8), with previously confirmed impaired function, and Vγ9Vδ2-T cells from HCs (n = 4) were sorted by FACS and subsequently cultured for 2 weeks with phosphoantigen-expressing moDCs in the presence of IL-7 and IL-15. Alternatively, Vγ9Vδ2-T cells (CLL, n = 4; HC, n = 4) were generated from CD19-depleted PBMCs by culture in the presence of ABPs and IL-2 for 2 weeks. (A) Expansion factor of Vγ9Vδ2-T cells calculated by dividing the amount of Vγ9Vδ2-T cells after a 2-week culture with allogeneic HC-derived moDCs (Allo-moDC) or autologous moDCs (Auto-moDC) by the number of Vγ9Vδ2-T cells at the start of culture. Cytokine production (B) and CD107a expression (C) by CLL-derived and HC-derived Vγ9Vδ2-T cells after culture with autologous moDCs. Vγ9Vδ2-T cells were cocultured with ABP-pretreated Daudi cells for 16 to 18 hours, and brefeldin A and GolgiStop were added during the last 6 hours of coculture. Granzyme B (D) and CD160 (F) expression on Vγ9Vδ2-T cells before and after culture with autologous moDCs. (E) Distribution of differentiation subsets within Vγ9Vδ2-T cells after culture with autologous moDCs based on CD27 and CD45RA expression. Data are mean and SEM. **P < .01, paired t test (D,F). TCM, CD27+CD45RA−; TEM, CD27−CD45RA−; TEMRA, CD27−CD45RA+; TN, CD27+CD45RA+.
Vγ9Vδ2-T–cell dysfunction is reversible upon ex vivo activation and expansion. Vγ9Vδ2-T cells from CLL patients (n = 8), with previously confirmed impaired function, and Vγ9Vδ2-T cells from HCs (n = 4) were sorted by FACS and subsequently cultured for 2 weeks with phosphoantigen-expressing moDCs in the presence of IL-7 and IL-15. Alternatively, Vγ9Vδ2-T cells (CLL, n = 4; HC, n = 4) were generated from CD19-depleted PBMCs by culture in the presence of ABPs and IL-2 for 2 weeks. (A) Expansion factor of Vγ9Vδ2-T cells calculated by dividing the amount of Vγ9Vδ2-T cells after a 2-week culture with allogeneic HC-derived moDCs (Allo-moDC) or autologous moDCs (Auto-moDC) by the number of Vγ9Vδ2-T cells at the start of culture. Cytokine production (B) and CD107a expression (C) by CLL-derived and HC-derived Vγ9Vδ2-T cells after culture with autologous moDCs. Vγ9Vδ2-T cells were cocultured with ABP-pretreated Daudi cells for 16 to 18 hours, and brefeldin A and GolgiStop were added during the last 6 hours of coculture. Granzyme B (D) and CD160 (F) expression on Vγ9Vδ2-T cells before and after culture with autologous moDCs. (E) Distribution of differentiation subsets within Vγ9Vδ2-T cells after culture with autologous moDCs based on CD27 and CD45RA expression. Data are mean and SEM. **P < .01, paired t test (D,F). TCM, CD27+CD45RA−; TEM, CD27−CD45RA−; TEMRA, CD27−CD45RA+; TN, CD27+CD45RA+.
After autologous moDC activation, the number of cells that produced IFN-γ, TNF-α, or IL-4 in response to Daudi cells was equivalent in CLL- and HC-derived Vγ9Vδ2-T cells (Figure 5B). Moreover, the reductions in degranulating CLL-derived Vγ9Vδ2-T cells (Figure 5C) and lysis of Daudi cells (supplemental Figure 4C) were no longer observed after autologous moDC activation. In line with this, granzyme B levels in Vγ9Vδ2-T cells increased during ex vivo activation and did not differ between HC- and CLL-derived Vγ9Vδ2-T cells (Figure 5D). To assess the long-term potential of expanded Vγ9Vδ2-T cells, the differentiation status after ex vivo activation was assessed. The Vγ9Vδ2-T cells were primarily differentiated toward a memory phenotype after moDC activation (Figure 5E). Moreover, there was a trend toward diminished CD160 expression on HC- and CLL-derived Vγ9Vδ2-T cells after ex vivo activation (Figure 5F).
Because the generation of moDCs complicates this ex vivo activation method, an expansion protocol based on previous clinical trials was also tested.28,30,45,46 In short, PBMCs were depleted, using magnetic bead depletion, from B cells and CLL cells and subsequently cultured in the presence of ABPs and IL-2 for 2 weeks. Using this protocol, we could generate functional Vγ9Vδ2-T cells from HCs and CLL patients (supplemental Figure 4). Yet, the expansion of the Vγ9Vδ2-T cells was inferior in comparison with the moDC-based protocol (Figure 5A).
In conclusion, ex vivo expansion of CLL-derived Vγ9Vδ2-T cells using a clinically feasible activation method resulted in fully functional Vγ9Vδ2-T cells with a memory phenotype.
The transcriptional profile of Vγ9Vδ2-T cells from CLL patients is globally altered
To gain more mechanistic insight into the observed differences between Vγ9Vδ2-T cells from HCs and CLL patients before activation, we performed RNA sequencing analyses on Vγ9Vδ2-T cells directly after thawing and after autologous moDC-based activation.
Although there was considerable interdonor variation, as could be expected with using primary T cells, principal component analysis showed that Vγ9Vδ2-T cells from CLL patients cluster apart from Vγ9Vδ2-T cells from HCs (Figure 6A). Ex vivo activation had a strong effect on the transcriptional profile of the Vγ9Vδ2-T cells, and Vγ9Vδ2-T cells from CLL patients and HCs clustered together after ex vivo activation. Thus, transcriptomics fitted with the previously observed altered function of Vγ9Vδ2-T cells from CLL patients in comparison with HC Vγ9Vδ2-T cells, which was no longer the case after ex vivo activation.
![Figure 6. Altered Vγ9Vδ2-T–cell transcriptional profile in CLL patients. RNA sequencing was performed on paired Vγ9Vδ2-T cells from 4 CLL patients and 4 HCs, directly after thawing and after autologous moDC-based expansion. (A) Multidimensional scaling plot of gene-expression data. Each circle represents an individual donor. (B) Number of genes differentially expressed (false discovery rate < 0.05) in the indicated comparisons. (C) Gene set enrichment analysis of CLL vs HC Vγ9Vδ2-T cells, before (x-axis) and after (y-axis) ex vivo expansion. Each circle represents a gene set [derived from the Hallmark (H) or BioCarta (B) gene sets from the Molecular Signatures Database or self-generated gene sets]. Gene sets differentially expressed (P < .01) before, but not after (P > .1), expansion are highlighted in orange and purple. (D) Gene set enrichment analysis of CLL vs HC Vγ9Vδ2-T cells directly prior to ex vivo expansion. The red dashed line indicates P = .05. Also shown is whether each gene set is up- or downregulated in CLL in comparison with HC Vγ9Vδ2-T cells. (E-F) The gene expression of CLL Vγ9Vδ2-T cells was compared with HC Vγ9Vδ2-T cells. Each circle represents an individual gene. Genes related to synapse and adhesion (E) or inhibitory and exhaustion (F) molecules are highlighted in red. Genes with a P value <.05 and/or a fold change ≤ −1.75 or ≥1.75 are annotated. FC, fold change; NK, natural killer cell.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/132/21/10.1182_blood-2017-12-822569/5/m_blood822569f6.png?Expires=1769094251&Signature=QzgKYto1CwMvdekBPUuZSU-ThVJW7~3TKGH11B2nMt7CtxchctpQ7h-VKiJoBnNPzxHa0~6j1otWIyfxiC54A~kpQfwuKqiF0~UFaYUWfnpZ5-R6T~FLTmR-DFZcniiGCZc~uljt3~BGjcflPtOH703G-kA9KoR4W5uyYulV2bBaAcTzL40M3yB4yw7oOTzyQGCN0fuOh6mKTj6RO5ZHTe~SIkk0-zZe9ictJrhmeFUBFxFU7QzDMajfiTDUuntXXHeFd8aTJrCdA3dSsA1SJABwMDtfRTntoUAYdcyD7GrV~qdoTUvJU8TvvMzTcQFIQM~~k1XzZ99EKVltXQd0zg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Altered Vγ9Vδ2-T–cell transcriptional profile in CLL patients. RNA sequencing was performed on paired Vγ9Vδ2-T cells from 4 CLL patients and 4 HCs, directly after thawing and after autologous moDC-based expansion. (A) Multidimensional scaling plot of gene-expression data. Each circle represents an individual donor. (B) Number of genes differentially expressed (false discovery rate < 0.05) in the indicated comparisons. (C) Gene set enrichment analysis of CLL vs HC Vγ9Vδ2-T cells, before (x-axis) and after (y-axis) ex vivo expansion. Each circle represents a gene set [derived from the Hallmark (H) or BioCarta (B) gene sets from the Molecular Signatures Database or self-generated gene sets]. Gene sets differentially expressed (P < .01) before, but not after (P > .1), expansion are highlighted in orange and purple. (D) Gene set enrichment analysis of CLL vs HC Vγ9Vδ2-T cells directly prior to ex vivo expansion. The red dashed line indicates P = .05. Also shown is whether each gene set is up- or downregulated in CLL in comparison with HC Vγ9Vδ2-T cells. (E-F) The gene expression of CLL Vγ9Vδ2-T cells was compared with HC Vγ9Vδ2-T cells. Each circle represents an individual gene. Genes related to synapse and adhesion (E) or inhibitory and exhaustion (F) molecules are highlighted in red. Genes with a P value <.05 and/or a fold change ≤ −1.75 or ≥1.75 are annotated. FC, fold change; NK, natural killer cell.
Altered Vγ9Vδ2-T–cell transcriptional profile in CLL patients. RNA sequencing was performed on paired Vγ9Vδ2-T cells from 4 CLL patients and 4 HCs, directly after thawing and after autologous moDC-based expansion. (A) Multidimensional scaling plot of gene-expression data. Each circle represents an individual donor. (B) Number of genes differentially expressed (false discovery rate < 0.05) in the indicated comparisons. (C) Gene set enrichment analysis of CLL vs HC Vγ9Vδ2-T cells, before (x-axis) and after (y-axis) ex vivo expansion. Each circle represents a gene set [derived from the Hallmark (H) or BioCarta (B) gene sets from the Molecular Signatures Database or self-generated gene sets]. Gene sets differentially expressed (P < .01) before, but not after (P > .1), expansion are highlighted in orange and purple. (D) Gene set enrichment analysis of CLL vs HC Vγ9Vδ2-T cells directly prior to ex vivo expansion. The red dashed line indicates P = .05. Also shown is whether each gene set is up- or downregulated in CLL in comparison with HC Vγ9Vδ2-T cells. (E-F) The gene expression of CLL Vγ9Vδ2-T cells was compared with HC Vγ9Vδ2-T cells. Each circle represents an individual gene. Genes related to synapse and adhesion (E) or inhibitory and exhaustion (F) molecules are highlighted in red. Genes with a P value <.05 and/or a fold change ≤ −1.75 or ≥1.75 are annotated. FC, fold change; NK, natural killer cell.
Increased expression of 104 genes was observed in CLL Vγ9Vδ2-T cells in comparison with HC Vγ9Vδ2-T cells prior to expansion (Figure 6B). The expression of considerably more genes (n = 501) was decreased in these CLL Vγ9Vδ2-T cells. In line with principal component analysis, no genes were differentially expressed between CLL and HC Vγ9Vδ2-T cells after ex vivo activation.
We hypothesized that the most relevant candidate gene sets underlying our functional observations would be differentially expressed between CLL and HC Vγ9Vδ2-T cells before ex vivo activation but comparably expressed after ex vivo activation (Figure 6C). Gene set enrichment analysis indicated that the concerned gene sets were primarily immune related and included TNF-α signaling via NF-κB and adhesion and diapedesis of lymphocytes.
Next, we compared the expression levels of selected genes hypothesized to be most relevant to Vγ9Vδ2-T–cell function (supplemental Table 2) between Vγ9Vδ2-T cells from CLL patients and HCs. Although the statistical power was limited, these data point to differential expression of genes that relate to T-cell memory, inhibition and exhaustion, costimulation, and synapse formation and adhesion (Figure 6D). Although genes involved in costimulation and memory were differentially expressed by CLL Vγ9Vδ2-T cells before activation, the same differences persisted after ex vivo activation (supplemental Figure 5A-C), making these unlikely candidates to explain our functional observations. We then zoomed in on genes related to synapse formation and adhesion, most of which were downregulated in CLL Vγ9Vδ2-T cells compared with HC Vγ9Vδ2-T cells before ex vivo activation (Figure 6E) but not after ex vivo activation (supplemental Figure 5F). Although there were some exceptions, genes attributed to T-cell inhibition and exhaustion were generally upregulated in CLL Vγ9Vδ2-T cells, most clearly before ex vivo activation (Figure 6F).
Thus, RNA sequencing confirmed that Vγ9Vδ2-T cells from CLL patients have a transcriptional profile that is distinct from HC Vγ9Vδ2-T cells and implicated impaired synapse formation and increased inhibitory molecules as conceivable contributors to compromised Vγ9Vδ2-T–cell function in CLL patients.
Ibrutinib promotes TH1 skewing of Vγ9Vδ2-T cells
The BTK inhibitor ibrutinib not only targets malignant B cells, it also skews αβ T cells toward a tumor-suppressive TH1 phenotype through inhibition of ITK.47 Whether ITK is expressed and exerts a similar role in Vγ9Vδ2-T cells has not been studied.
CLL-derived Vγ9Vδ2-T cells that were pretreated with ibrutinib produced significantly more TNF-α upon coculture with ABP-treated Daudi cells (Figure 7A). In contrast, IL-4–producing Vγ9Vδ2-T–cell numbers declined after ibrutinib treatment. Ibrutinib treatment did not impair the degranulation of Vγ9Vδ2-T cells (Figure 7B). To study whether effects of ibrutinib indeed depended on inhibition of targets other than BTK, Vγ9Vδ2-T cells from CLL patients were pretreated with the highly BTK-specific inhibitor CC-292.48 In contrast to ibrutinib, CC-292 did not alter cytokine production by Vγ9Vδ2-T cells (Figure 7C).
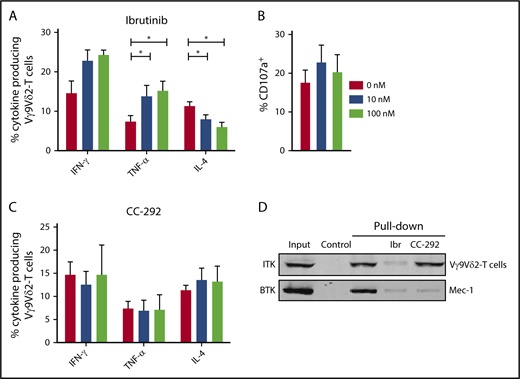
Ibrutinib promotes TH1 phenotype in Vγ9Vδ2-T cells. (A-B) CD19-depleted PBMCs were treated with 0, 10, or 100 nM ibrutinib for 30 minutes and subsequently cocultured with ABP-pretreated (25 μM pamidronate for 2 hours) Daudi cells. During the last 6 hours of coculture, brefeldin A and GolgiStop were added to measure cytokine production (A) and CD107a expression (B) in Vγ9Vδ2-T cells (n = 10). (C) Cytokine production as in (A) after pretreatment with 0, 10, or 100 nM CC-292 (n = 6). Data are mean and SEM. (D) Pull-down with biotinylated ibrutinib coupled to avidin agarose; uncoupled avidin agarose was used as a control. Lysates from healthy Vγ9Vδ2-T cells or control Mec-1 B cells were treated with 1 μM ibrutinib or 1 μM CC-292 before pull-down. Representative result of 3 donors in 2 independent experiments is shown. *P < .05, 1-way ANOVA, followed by the Dunnett post hoc test.
Ibrutinib promotes TH1 phenotype in Vγ9Vδ2-T cells. (A-B) CD19-depleted PBMCs were treated with 0, 10, or 100 nM ibrutinib for 30 minutes and subsequently cocultured with ABP-pretreated (25 μM pamidronate for 2 hours) Daudi cells. During the last 6 hours of coculture, brefeldin A and GolgiStop were added to measure cytokine production (A) and CD107a expression (B) in Vγ9Vδ2-T cells (n = 10). (C) Cytokine production as in (A) after pretreatment with 0, 10, or 100 nM CC-292 (n = 6). Data are mean and SEM. (D) Pull-down with biotinylated ibrutinib coupled to avidin agarose; uncoupled avidin agarose was used as a control. Lysates from healthy Vγ9Vδ2-T cells or control Mec-1 B cells were treated with 1 μM ibrutinib or 1 μM CC-292 before pull-down. Representative result of 3 donors in 2 independent experiments is shown. *P < .05, 1-way ANOVA, followed by the Dunnett post hoc test.
To test whether ITK was bound by ibrutinib in Vγ9Vδ2-T cells, we performed competitive pull-down experiments utilizing biotinylated ibrutinib coupled to avidin-agarose. Specific binding of BTK to biotinylated ibrutinib was confirmed in the CLL-derived Mec-1 cell line, which could be reverted by ibrutinib or CC-292 pretreatment (Figure 7D). ITK was bound by biotinylated ibrutinib in Vγ9Vδ2-T cells. Pretreatment with ibrutinib, but not with CC-292, blocked ITK binding.
Taken together, these experiments demonstrate that ibrutinib, but not CC-292, promotes a TH1 phenotype in Vγ9Vδ2-T cells from CLL patients that is likely mediated by ITK inhibition.
Discussion
Vγ9Vδ2-T cells are attractive effector cells for immunotherapy that are capable of major histocompatibility complex–independent recognition of malignant cells. In line with the cytotoxic capacity of Vγ9Vδ2-T cells toward other malignant B cells,18,21-24 we demonstrate Vγ9Vδ2-T–cell cytotoxicity toward CLL cells.
However, clinical application of autologous Vγ9Vδ2-T cells seems hampered by the observed dysfunction in Vγ9Vδ2-T cells. Indeed, CLL cells have been found to have an active immunosuppressive function.49,50 In agreement with these findings, we show here that CLL cells also induce dysfunction in healthy donor–derived Vγ9Vδ2-T cells. Although the expression of exhaustion markers, especially PD-1, correlates with αβ T-cell dysfunction in CLL,13 Vγ9Vδ2-T cells do not express increased transcriptional or protein levels of PD-1. On the other hand, CD160 expression was upregulated on Vγ9Vδ2-T cells from CLL patients, and genes related to T-cell exhaustion were generally expressed at higher transcriptional levels in CLL Vγ9Vδ2-T cells. Although the functional role of CD160 in γδ T cells has only been described in the context of CD3 stimulation,51 it is plausible that CD160 has an inhibitory role in Vγ9Vδ2-T cells, as was described in αβ T cells.52 Gene-expression profiling also implicated alterations in synapse formation in the observed cytotoxic impairment, as has been demonstrated for αβ T cells in CLL.14 Vγ9Vδ2-T–cell anergy can arise as a result of chronic phosphoantigen overstimulation.53,54 A similar mechanism in CLL is supported by our observation that naive Vγ9Vδ2-T cells are lost, and dysfunction increases with disease stage, as well as that Vγ9Vδ2-T–cell numbers were increased in advanced-stage CLL in comparison with monoclonal B-cell lymphocytosis and Rai stage 0 disease.55 The possibility of chronic overstimulation of Vγ9Vδ2-T cells as the result of a hyperactive mevalonate pathway in U-CLL cells was proposed previously.31
We explored several approaches to improve the function of CLL-derived Vγ9Vδ2-T cells. Pharmacological enhancement of phosphoantigen levels is clinically feasible, and in vivo induction of Vγ9Vδ2-T–cell proliferation using administration of ABPs or synthetic phosphoantigens and IL-2 has been achieved in solid25-27 and lymphoid24 malignancies. However, ABP treatment only led to a modest lysis-sensitizing effect of primary CLL cells in vitro, which might be explained by overactivity of the mevalonate pathway in CLL.
Next, we examined ex vivo activation of Vγ9Vδ2-T cells in light of adoptive-transfer strategies. Clinical trials have demonstrated safety28-30 and efficacy30,45,56,57 of ex vivo–expanded Vγ9Vδ2-T–cell administration. In line with our data, other investigators have shown that sufficient Vγ9Vδ2-T–cell numbers can be achieved by expansion using coculture with moDCs,58 as well as through adding synthetic phosphoantigens or ABPs to PBMCs.56 Moreover, we demonstrate that it is possible to generate Vγ9Vδ2-T cells with regained functionality in an autologous ex vivo culture system that is clinically feasible. The function of Vγ9Vδ2-T cells improved for all patients tested, although the number of patients tested was limited. Activation with moDCs was performed in the presence of IL-15, which was previously shown to be superior to IL-2 for induction of sustained proliferation, TH1 skewing, and degranulation.46,59 Most Vγ9Vδ2-T cells had a memory phenotype after ex vivo activation, which is favorable in light of long-term functionality.60-62
Finally, we investigated combination strategies with novel BTK inhibitors, which allow for selective elimination of CLL cells while largely sparing T cells. In fact, ibrutinib has been shown to increase T-cell numbers and reduce the immunosuppressive features of CLL,63,64 conceivably favoring subsequent autologous T-cell therapy. Specifically, ibrutinib promotes a favorable antitumor TH1 profile in Vγ9Vδ2-T cells derived from CLL patients, similar to what was previously shown for CD4+ T cells.47,63 Because TH1 skewing did not occur with CC-292, the effect is likely mediated by inhibition of ITK, as was supported by our pull-down experiments.48,65 Hence, ibrutinib may be a suitable candidate to combine with autologous Vγ9Vδ2-T-cell–based therapy.
In conclusion, Vγ9Vδ2-T cells are cytotoxic toward CLL cells, and dysfunction of autologous Vγ9Vδ2-T cells is fully reversible upon ex vivo expansion. Because autologous Vγ9Vδ2-T cells can be expanded to high numbers, and ibrutinib stimulates an antitumor TH1 pattern, further exploration of combining Vγ9Vδ2-T–cell adoptive transfer with ibrutinib treatment is justified.
Sequence data have been deposited in the European Genome-phenome Archive under accession number EGAS00001003193.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank the patients and healthy donors for their blood donations.
This work was supported by an AMC/Ph.D. Scholarship (I.d.W.) and NWO/ZonMw VIDI grant (A.P.K.).
Authorship
Contribution: I.d.W. designed and performed experiments, analyzed data. and wrote the manuscript; T.H., R.L., S.E., and R.C.G.d.B. performed experiments and reviewed the manuscript; A.J. and P.D.M. analyzed data and reviewed the manuscript; N.L. and M.v.d.S. provided biotinylated ibrutinib; L.M.F. and M.-D.L. provided patient samples and reviewed the manuscript; E.B.M.R., I.J.M.t.B., E.E., S.H.T., and T.D.d.G. contributed to the design of experiments and reviewed the manuscript; and H.J.v.d.V. and A.P.K. designed the study and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Arnon P. Kater, Department of Hematology, Amsterdam UMC, Meibergdreef 9, 1105AZ Amsterdam, The Netherlands; e-mail: a.p.kater@amc.nl.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal