

Key Points
TNF-α from host macrophages and TNF-αRs expressed on donor T cells are critical in the pathogenesis of murine immune-mediated BM failure.
AA patients have higher frequencies of TNF-α–producing CD16+CD68+ macrophages in the BM than healthy controls.

Abstract
Interferon-γ (IFN-γ) and tumor necrosis factor-α (TNF-α) have been implicated historically in the immune pathophysiology of aplastic anemia (AA) and other bone marrow (BM) failure syndromes. We recently defined the essential roles of IFN-γ produced by donor T cells and the IFN-γ receptor in the host in murine immune-mediated BM failure models. TNF-α has been assumed to function similarly to IFN-γ. We used our murine models and mice genetically deficient in TNF-α or TNF-α receptors (TNF-αRs) to establish an analogous mechanism. Unexpectedly, infusion of TNF-α−/− donor lymph node (LN) cells into CByB6F1 recipients or injection of FVB LN cells into TNF-αR−/− recipients both induced BM failure, with concurrent marked increases in plasma IFN-γ and TNF-α levels. Surprisingly, in TNF-α−/− recipients, BM damage was attenuated, suggesting that TNF-α of host origin was essential for immune destruction of hematopoiesis. Depletion of host macrophages before LN injection reduced T-cell IFN-γ levels and reduced BM damage, whereas injection of recombinant TNF-α into FVB-LN cell-infused TNF-α−/− recipients increased T-cell IFN-γ expression and accelerated BM damage. Furthermore, infusion of TNF-αR−/− donor LN cells into CByB6F1 recipients reduced BM T-cell infiltration, suppressed T-cell IFN-γ production, and alleviated BM destruction. Thus, TNF-α from host macrophages and TNF-αR expressed on donor effector T cells were critical in the pathogenesis of murine immune-mediated BM failure, acting by modulation of IFN-γ secretion. In AA patients, TNF-α–producing macrophages in the BM were more frequent than in healthy controls, suggesting the involvement of this cytokine and these cells in human disease.
Introduction
Aplastic anemia (AA) is a bone marrow (BM) failure syndrome characterized by pancytopenia and BM hypoplasia, in most patients due to immune destruction of hematopoietic stem and progenitor cells (HSPCs) by autoreactive T cells.1,2 Upregulation of type I cytokines interferon-γ (IFN-γ) and tumor necrosis factor-α (TNF-α) has been implicated as a critical molecular event in the destruction of BM HSPCs. The inflammatory cytokine IFN-γ is important in both innate and adaptive immunity against viral, bacterial, and protozoal infections because it functions as the primary activator of macrophages, natural killer cells, and neutrophils under these circumstances. The roles of IFN-γ in AA and immune-mediated BM failure have been well documented: (1) IFN-γ inhibits the proliferation of human progenitor cells in vitro3-5 ; (2) overexpression of IFN-γ in BM cells and T cells is associated with immune-mediated BM failure6,7 ; (3) upregulation of T-bet and other gene elements in the IFN-γ–signaling pathway is observed in untreated AA patients8 ; (4) IFN-γ stimulates Fas expression on HSPCs in the BM, facilitating destruction by activated T cells through the Fas/FasL apoptosis pathway.9,10
TNF-α also is critical in systemic inflammation and is a potent inducer of apoptotic cell death. Increased TNF-α production has been implicated in the development of diabetes, septic shock, tumorigenesis, cardiovascular diseases, rheumatoid arthritis, and inflammatory bowel disease,11 and targeting TNF-α has emerged as useful treatment of many of these diseases.12 For example, TNF-α antagonists are effective in rheumatoid arthritis.13 However, occasional paradoxical instances of lupus-like syndrome and skin lesions, as well as AA, neutropenia, and thrombocytopenia, have been reported in patients receiving anti-TNF therapies.14-18 TNF-α is implicated in the pathophysiology of hematologic diseases, including anemia and myelodysplasia, in which TNF-α appears as an important negative regulator of hematopoiesis.7,13,19,20 Although upregulation of TNF-α in T cells and TNF-α receptors (TNF-αRs) on BM CD34+ cells has been described in patients with AA,21,22 the precise role of TNF-α, and especially its functional mechanisms and relationship to IFN-γ, in immune-mediated BM failure have not been well characterized.
We have modeled human AA in mice by adaptation of historic “runt disease,” in which infusion of lymph node (LN) cells into recipients mismatched at major histocompatibility complex23,24 or minor histocompatibility antigen25 loci produces severe pancytopenia and BM failure. Using these models, we recently reexamined the role of the IFN-γ/IFN-γ receptor signaling pathway in BM failure: IFN-γ−/− donor T cells failed to induce BM destruction; IFN-γ receptor−/− recipient mice did not develop marrow failure when infused with major histocompatibility complex–mismatched FVB/N (FVB) LN cells. In these models, T cells cause BM destruction by activation of the IFN-γ/IFN-γ receptor signaling pathway.10 In the present study, we used the same model systems and mice deficient in TNF-α or TNF-αR to study the specific roles of TNF-α/TNF-αR in BM failure. Our unexpected observations reveal a complex interactive loop involving production of TNF-α by macrophages, engagement of TNF-α with TNF-αR on effector T cells, and production of IFN-γ by activated effector T cells. We also examined marrow samples from AA patients and found increased macrophages with high levels of intracellular TNF-α compared with healthy individuals.
Materials and methods
Animals and induction of BM failure
Inbred C57BL/6 (B6) and FVB, induced mutants B6.129S-Tnftm1Gkl/J (TNF-α−/−), B6.129-Tnfrsf1atm1Mak/J (TNFrsf1a−/−), B6.129S2-Tnfrsf1btm1Mwm/J (TNFrsf1b−/−), and B6.129S-Tnfrsf1atm1lmx Tnfrsf1btm1lmx/J (TNFrsf1a−/−1b−/−), and hybrid (BALB/cBy×B6)F1 (CByB6F1) mice were all purchased from The Jackson Laboratory (Bar Harbor, ME) and were bred and maintained in National Institutes of Health animal facilities with standard care and nutrition. Animals were used at 8 to 16 weeks of age. All animal studies were approved by the Animal Care and Use Committee of the National Heart, Lung, and Blood Institute.
BM failure was induced by allogeneic LN-cell infusion: LN cells from B6-based (wild-type [WT], TNF-α−/−, or TNFrsf1a−/−1b−/−) donors were injected into CByB6F1 recipients preirradiated with 5 Gy of total-body irradiation (TBI) to analyze animals at day 13,23 or LN cells from FVB donors were injected into B6-based (WT, TNF-α−/−, TNFrsf1a−/−, TNFrsf1b−/−, or TNFrsf1a−/−1b−/−) recipients preirradiated with 6.5 Gy TBI to analyze recipients at day 10.24
Recombinant TNF-α injection and macrophage depletion in vivo
Recombinant murine TNF-α was obtained from PeproTech (Rocky Hill, NJ), and administrated through IV injection at 100 ng per mouse once daily for 1 week following BM failure induction. Control mice were injected with the same volume of normal saline at the same time.
Depletion of macrophages in vivo was performed as described previously, with minor modifications.26,27 Mice were injected retro-orbitally with 200 to 300 μL of a clodronate-loaded liposome suspension (Clodrosome; Encapsula NanoSciences, Nashville, TN) every other day for a total of 4 doses in the week prior to BM failure induction. Control mice were injected with 200 to 300 μL of phosphate-buffered saline–loaded liposomes (Encapsome) at the same time points.
Details for blood counts, cell staining and flow cytometry, cytokine measurement, cell culture in vitro, and polymerase chain reaction (PCR) array are included in supplemental Methods (available on the Blood Web site).
Human samples
Heparinized whole peripheral blood and BM were collected from patients and healthy subjects after informed consent was obtained, in accordance with the Declaration of Helsinki28 and protocols approved by the National Heart, Lung, and Blood Institute Institutional Review Board (National Institutes of Health, Bethesda, MD) (see supplemental Table 1 for clinical characteristics). All patients received a diagnosis of severe AA according to the International Study of Aplastic Anemia and Agranulocytosis29 and to the criteria of Camitta.30 At the time of blood or BM sampling, none of the patients had received specific therapy.
Statistics
Data obtained from complete blood count, BM cell counting, and flow cytometry were analyzed by the unpaired Student t test, variance analyses, and multiple comparisons using GraphPad Prism and JMP statistical discovery software, respectively. Data are presented as means with standard deviations. Statistical significance was declared at P < .05.
Results
Lymphocytes genetically deficient in TNF-α induce immune BM failure in the mice
As T cells, especially CD8+ T cells, are the effectors responsible for BM destruction, we first tested whether TNF-α in effector T cells was essential for induction of immune-mediated BM failure. We infused LN cells from B6 WT or TNF-α−/− donors into preirradiated CByB6F1 recipients (Figure 1A). Injection of either B6 WT or TNF-α−/− LN cells caused severe BM failure in recipient mice, relative to TBI-only controls, evidenced by: empty BM cavity (Figure 1B); reduced numbers of white blood cells (WBCs), platelets, and total BM cells (Figure 1C); increased Fas expression on residual BM cells (Figure 1D); reduced frequencies of total colony-forming unit (CFU) and subcomponents CFU-granulocyte, macrophage (CFU-GM), CFU-granulocyte (CFU-G), and CFU-macrophage (CFU-M) (Figure 1E); and increased plasma levels of IFN-γ and TNF-α (Figure 1F). Surprisingly, LN cells deficient in TNF-α were fully functional in induction of immune-mediated BM failure, in contrast to LN cells deficient in IFN-γ production.10
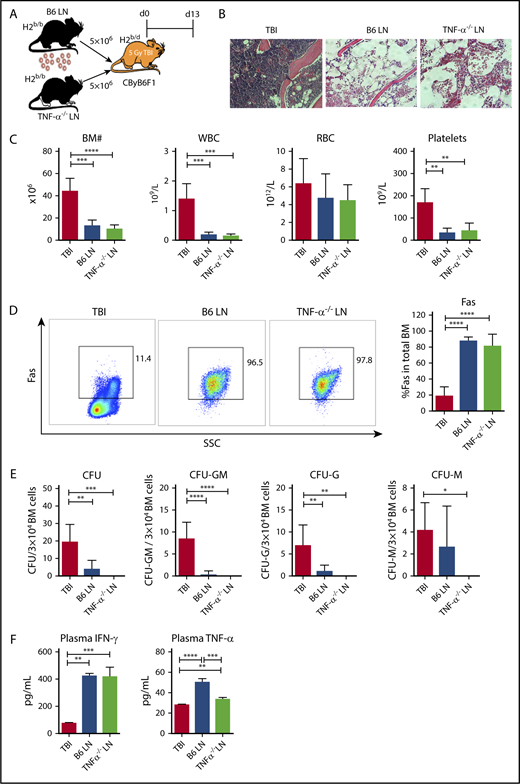
TNF-α deficiency in donor effector cells does not block T-cell–mediated BM destruction. (A) CByB6F1 mice were irradiated with 5 Gy TBI without (TBI; n = 4) or with infusion of 5 × 106 LN cells from WT C57BL/6 (B6 LN; n = 5) or TNF-α−/− (TNF-α−/− LN; n = 5) donors to induce BM failure. Animals were euthanized and evaluated at day 13. (B) Histology of sternums from CByB6F1 mice that received TBI, TBI plus B6 LN, or TBI plus TNF-α−/− LN treatments shown as representative images (original magnification ×200; hematoxylin and eosin stain). (C) CByB6F1 mice that received LN infusion from either WT B6 or TNF-α−/− donors developed BM failure showing reduction in BM cells, WBCs, and platelets. (D) CByB6F1 mice that received B6 LN or TNF-α−/− LN infusion expressed high levels of Fas on whole BM cells. (E) BM cells from CByB6F1 mice that received B6 LN or TNF-α−/− LN infusion showed reduced total CFU, CFU-GM, CFU-G, and CFU-M, relative to control mice treated with TBI only. (F) CByB6F1 mice that received B6 LN or TNF-α−/− LN infusion had high levels of IFN-γ and TNF-α in the plasma. *P < .05; **P < .01; ***P < .001; ****P < .0001. SSC, side scatter.
TNF-α deficiency in donor effector cells does not block T-cell–mediated BM destruction. (A) CByB6F1 mice were irradiated with 5 Gy TBI without (TBI; n = 4) or with infusion of 5 × 106 LN cells from WT C57BL/6 (B6 LN; n = 5) or TNF-α−/− (TNF-α−/− LN; n = 5) donors to induce BM failure. Animals were euthanized and evaluated at day 13. (B) Histology of sternums from CByB6F1 mice that received TBI, TBI plus B6 LN, or TBI plus TNF-α−/− LN treatments shown as representative images (original magnification ×200; hematoxylin and eosin stain). (C) CByB6F1 mice that received LN infusion from either WT B6 or TNF-α−/− donors developed BM failure showing reduction in BM cells, WBCs, and platelets. (D) CByB6F1 mice that received B6 LN or TNF-α−/− LN infusion expressed high levels of Fas on whole BM cells. (E) BM cells from CByB6F1 mice that received B6 LN or TNF-α−/− LN infusion showed reduced total CFU, CFU-GM, CFU-G, and CFU-M, relative to control mice treated with TBI only. (F) CByB6F1 mice that received B6 LN or TNF-α−/− LN infusion had high levels of IFN-γ and TNF-α in the plasma. *P < .05; **P < .01; ***P < .001; ****P < .0001. SSC, side scatter.
Disruption of TNF-α/TNF-αR signaling in recipient mice does not attenuate BM failure
To test whether T-cell–mediated BM destruction was mediated through the TNF-α/TNF-αR signaling pathways, we infused LN cells from FVB donors into preirradiated B6 WT mice as well as into B6 mice carrying TNF-αR gene deletions (TNFrsf1a−/−, TNFrsf1b−/−, and TNFrsf1a−/−1b−/−; supplemental Figure 1A). Injection of FVB LN cells induced severe BM failure in animals of all 3 gene-deletion genotypes (supplemental Figure 1B), and affected animals showed very low BM cells and platelets relative to TBI-only controls, as in B6 WT recipients (supplemental Figure 1C) (deletion of TNFrsf1b appeared to increase sensitivity to irradiation [supplemental Figure 1C]). Plasma concentrations of IFN-γ and TNF-α both were increased in all mice that received FVB LN cells, relative to TBI-only controls (supplemental Figure 1D). Thus, deletion of TNF-αR in recipient animals did not attenuate immune-mediated BM failure.
Immune-mediated BM failure is abrogated in TNF-α−/− recipient mice
Although absence of TNF-α in donor T cells and of TNF-αR in recipient cells did not abrogate BM failure in our model animals, in control experiments we serendipitously observed TNF-α−/− recipient mice to be resistant to T-cell–mediated BM destruction. We used the FVB→B6 BM failure model by injecting FVB LN cells into preirradiated B6 WT or TNF-α−/− recipients (Figure 2A). B6 WT manifested severe BM failure post-LN injection, but TNF-α−/− recipient mice were resistant to FVB LN cells: BM cellularity was unaffected and marrow structure was intact (Figure 2B); there were similar or higher levels of WBCs, red blood cells (RBCs), platelets in the blood; and total BM cell numbers were similar to TBI controls (Figure 2C). When BM cells were cultured in vitro in semisolid medium, colony formation by BM cells from TNF-α−/− recipient mice showed similar frequencies of total CFU and CFU-GM, CFU-G, and CFU-M as from TBI controls, and significantly more colonies compared with BM of B6 mice after LN infusion (Figure 2D). Additionally, there was a lower percentage of BM cells expressing Fas (Figure 2E), and lower levels of IFN-γ and TNF-α in blood plasma (Figure 2F) in TNF-α−/− recipient mice, when compared with TBI-only TNF-α−/− controls. Thus, there was little evidence of immune activation in TNF-α−/− recipient mice, in contrast to marked immune activation in B6 WT recipient mice, and we inferred that recipient animals with germline deletion of TNF-α were resistant to immune BM destruction.
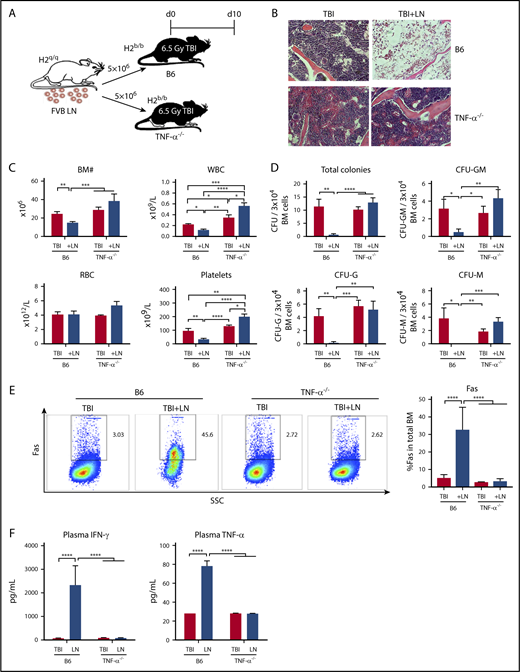
TNF-α–deficient mice are resistant to immune-mediated BM failure. (A) WT C57BL/6 (B6) mice or B6 mice carrying germline deletion of TNF-α (TNF-α−/−) were irradiated with 6.5 Gy TBI without (TBI) or with infusion of 5 × 106 LN cells (TBI+LN) from FVB donors to induce BM failure. Animals were evaluated at day 10. (B) Histology of sternums from B6 or TNF-α−/− mice that received TBI or TBI+LN infusion (original magnification ×200; hematoxylin and eosin stain). (C) B6 mice that received TBI+LN infusion (n = 10) showed declines in WBCs, platelets, and total BM cells relative to TBI controls (n = 8), whereas TNF-α−/− animals that received TBI+LN infusion (n = 5) showed no decline in blood or BM cells relative to TBI controls (n = 4). (D) TBI+LN infusion caused significant declines in BM total CFU, CFU-G, CFU-M, and CFU-GM in B6 mice, whereas no such decline was detected in TBI+LN-infused TNF-α−/− mice. (E) Fas expression on total BM cells was increased in B6, but not in TNF-α−/−, recipient mice following FVB LN-cell injection. (F) Blood plasma concentrations of IFN-γ and TNF-α increased drastically in B6, but not in TNF-α−/−, recipient mice following FVB LN-cell infusion, relative to their respective TBI controls. *P < .05; **P < .01; ***P < .001; ****P < .0001.
TNF-α–deficient mice are resistant to immune-mediated BM failure. (A) WT C57BL/6 (B6) mice or B6 mice carrying germline deletion of TNF-α (TNF-α−/−) were irradiated with 6.5 Gy TBI without (TBI) or with infusion of 5 × 106 LN cells (TBI+LN) from FVB donors to induce BM failure. Animals were evaluated at day 10. (B) Histology of sternums from B6 or TNF-α−/− mice that received TBI or TBI+LN infusion (original magnification ×200; hematoxylin and eosin stain). (C) B6 mice that received TBI+LN infusion (n = 10) showed declines in WBCs, platelets, and total BM cells relative to TBI controls (n = 8), whereas TNF-α−/− animals that received TBI+LN infusion (n = 5) showed no decline in blood or BM cells relative to TBI controls (n = 4). (D) TBI+LN infusion caused significant declines in BM total CFU, CFU-G, CFU-M, and CFU-GM in B6 mice, whereas no such decline was detected in TBI+LN-infused TNF-α−/− mice. (E) Fas expression on total BM cells was increased in B6, but not in TNF-α−/−, recipient mice following FVB LN-cell injection. (F) Blood plasma concentrations of IFN-γ and TNF-α increased drastically in B6, but not in TNF-α−/−, recipient mice following FVB LN-cell infusion, relative to their respective TBI controls. *P < .05; **P < .01; ***P < .001; ****P < .0001.
Depletion of recipient macrophages attenuates BM destruction
Different from IFN-γ/IFN-γ receptor signaling in murine BM destruction,10 TNF-α production in recipient animals (Figure 2) rather than in donor effector cells (Figure 1) appeared essential in eliciting the disease phenotypes; TNF-αR expression on recipient target cells was not required (supplemental Figure 1). Nevertheless, increased plasma levels of IFN-γ and TNF-α in the animals suffering BM failure (Figure 1F; supplemental Figure 1D; Figure 2F) suggested that interactions between the 2 cytokines might underlie the pathophysiology. As macrophages are the major source of TNF-α, we reasoned that macrophages producing TNF-α modulated T-cell IFN-γ production and mediated BM destruction. To test this idea, we depleted macrophages from CByB6F1 recipient mice with clodronate (Clodrosome) before attempting to induce BM failure (Figure 3A). In the mice with macrophage depletion, B6 LN-cell–mediated BM destruction was markedly attenuated (Figure 3B), evidenced by increased BM cell numbers, RBCs, and platelets, relative to controls (mice preinjected with phosphate-buffered saline–loaded liposomes Encapsome; Figure 3B). Depletion of macrophages inhibited BM infiltration of effector T cells, especially CD8+ T cells (Figure 3C), and reduced circulating IFN-γ and TNF-α (Figure 3D) in plasma as well as the proportions of IFN-γ–secreting CD4+ and CD8+ T cells in BM (Figure 3E).
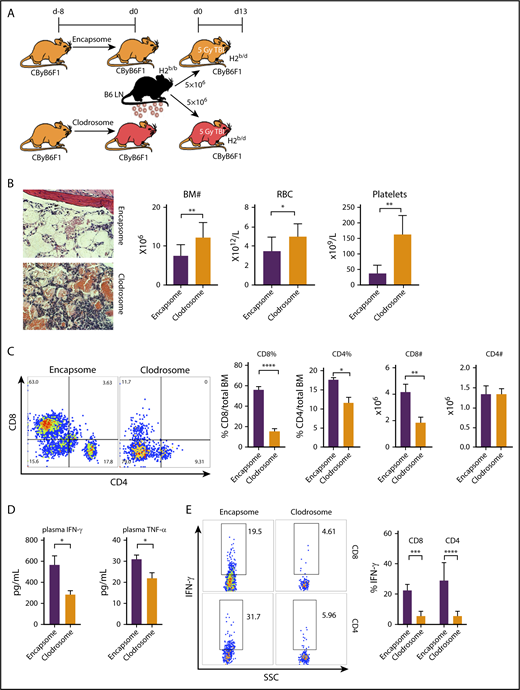
Macrophage depletion ameliorates BM failure. (A) Macrophages from CByB6F1 recipient mice were depleted with Clodrosome before induction of BM failure; Encapsome was used as vehicle control. Severity of BM failure was evaluated at day 13. (B) Macrophage depletion with Clodrosome (n = 9) attenuated immune-mediated BM failure in CByB6F1 mice with improved BM structure, and increased numbers of BM cells, RBCs, and platelets, when compared with mice treated with Encapsome as vehicle control (n = 5) without macrophage depletion (original magnification ×200; hematoxylin and eosin stain). (C) Macrophage depletion by Clodrosome suppressed expansion of T cells, especially CD8+ T cells, in the BM relative to vehicle control without macrophage depletion. (D) Macrophage depletion by Clodrosome reduced plasma levels of IFN-γ and TNF-α. (E) Macrophage depletion by Clodrosome suppressed IFN-γ levels in both CD8+ and CD4+ T cells in the BM. *P < .05; **P < .01; ***P < .001; ****P < .0001.
Macrophage depletion ameliorates BM failure. (A) Macrophages from CByB6F1 recipient mice were depleted with Clodrosome before induction of BM failure; Encapsome was used as vehicle control. Severity of BM failure was evaluated at day 13. (B) Macrophage depletion with Clodrosome (n = 9) attenuated immune-mediated BM failure in CByB6F1 mice with improved BM structure, and increased numbers of BM cells, RBCs, and platelets, when compared with mice treated with Encapsome as vehicle control (n = 5) without macrophage depletion (original magnification ×200; hematoxylin and eosin stain). (C) Macrophage depletion by Clodrosome suppressed expansion of T cells, especially CD8+ T cells, in the BM relative to vehicle control without macrophage depletion. (D) Macrophage depletion by Clodrosome reduced plasma levels of IFN-γ and TNF-α. (E) Macrophage depletion by Clodrosome suppressed IFN-γ levels in both CD8+ and CD4+ T cells in the BM. *P < .05; **P < .01; ***P < .001; ****P < .0001.
TNF-α administration accelerates BM destruction and stimulates T-cell IFN-γ secretion
To confirm the role of TNF-α and to elucidate TNF-α/IFN-γ interactions in immune-mediated BM failure, we infused FVB LN cells into preirradiated TNF-α−/− recipient mice with or without 1 week of daily recombinant murine TNF-α (Figure 4A). At day 10 after LN-cell infusion, TNF-α−/− recipients that had received both FVB LN cells and TNF-α by IV injection had poorer BM cell recovery and more severe BM damage than did TNF-α−/− mice that had received FVB LN cells without TNF-α (Figure 4B). By flow cytometry, there were higher proportions of IFN-γ–producing CD4+ and CD8+ T cells in BM in the TNF-α−/− mice treated with recombinant TNF-α, compared with controls (Figure 4C). Injection of recombinant TNF-α did not affect intracellular TNF-α levels in CD4+ and CD8+ T cells, confirming general deficiency of TNF-α production in TNF-α−/− mice (Figure 4D).

Exogenous TNF-α accelerates BM destruction and promotes T-cell IFN-γ secretion in TNF-α−/−mice. (A) TNF-α−/− mice received 6.5 Gy TBI and infusion of 5 × 106 FVB LN cells were either untreated (n = 4), or were injected with recombinant TNF-α protein at 100 ng per day IV for 7 days (n = 5). Mice were euthanized and analyzed on day 10. Injection of recombinant TNF-α to LN-cell–infused TNF-α−/− mice reduced BM cellularity (n = 4; original magnification ×200; hematoxylin and eosin stain) (B), significantly increased intracellular IFN-γ levels in BM CD4+ and CD8+ T cells (C), but did not change intracellular TNF-α levels in BM CD4+ and CD8+ T cells (D), when compared with LN-cell–infused TNF-α−/− mice without TNF-α injection. *P < .05; **P < .01.
Exogenous TNF-α accelerates BM destruction and promotes T-cell IFN-γ secretion in TNF-α−/−mice. (A) TNF-α−/− mice received 6.5 Gy TBI and infusion of 5 × 106 FVB LN cells were either untreated (n = 4), or were injected with recombinant TNF-α protein at 100 ng per day IV for 7 days (n = 5). Mice were euthanized and analyzed on day 10. Injection of recombinant TNF-α to LN-cell–infused TNF-α−/− mice reduced BM cellularity (n = 4; original magnification ×200; hematoxylin and eosin stain) (B), significantly increased intracellular IFN-γ levels in BM CD4+ and CD8+ T cells (C), but did not change intracellular TNF-α levels in BM CD4+ and CD8+ T cells (D), when compared with LN-cell–infused TNF-α−/− mice without TNF-α injection. *P < .05; **P < .01.
TNF-α modulates T-cell function through TNF-αR
Because depletion of macrophages reduced T-cell IFN-γ and attenuated BM failure in CByB6F1 mice upon B6 LN-cell infusion (Figure 3), whereas injection of TNF-α increased T-cell IFN-γ and accelerated BM destruction in TNF-α−/− mice following FVB LN-cell infusion (Figure 4), we hypothesized that TNF-α bound to TNF-αR on donor T cells and stimulated T-cell IFN-γ expression/secretion. TNF-α acts through at least 2 distinct TNF-αR, TNFrsf1a and TNFrsf1b. We tested our hypothesis by infusing LN cells from B6 WT or TNFrsf1a−/−1b−/− donors into preirradiated CByB6F1 recipients (Figure 5A). BM failure was attenuated in animals receiving TNFrsf1a−/−1b−/− LN cells compared with those receiving B6 WT LN cells, showing higher numbers of BM cells, WBCs, RBCs, and platelets (Figure 5B). In B6 LN-injected mice, there was marked T-cell infiltration of marrow, mainly by CD8+ T cells; the frequency of BM-infiltrating CD8+ T cells was much decreased in TNFrsf1a−/−1b−/− LN-injected mice, but the absolute numbers of CD8+ and CD4+ T cells were not changed (Figure 5C). The frequencies of IFN-γ–producing CD4+ and CD8+ T cells in the BM from TNFrsf1a−/−1b−/− LN-injected mice were significantly lower compared with B6 LN-injected mice (Figure 5D). When BM CD8+ T cells were tested for activation/differentiation based on expression of CD44 and CD62L, we found that >80% BM CD8+ T cells from B6 LN-injected mice were CD44+CD62L− effector memory cells and 14% were CD44+CD62L+ central memory cells (Figure 5E). In contrast, of BM CD8+ T cells from TNFrsf1a−/−1b−/− LN-injected mice, 50% had the effector memory phenotype and 40% displayed central memory phenotype, suggesting delayed CD8+ T-cell differentiation from central memory to effector memory (Figure 5E). Most BM CD4+ T cells from TNFrsf1a−/−1b−/− LN-infused mice were effector memory phenotype, as in B6 LN-infused control mice (data not shown). Consistent with reduced T-cell IFN-γ secretion and the attenuated BM destruction, recipients of TNFrsf1a−/−1b−/− LN cells also had lower Fas expression on residual BM cells (Figure 5F). When we included individual TNF-αR knockout donor LN cells in the experiment, BM and peripheral blood cells appeared to be better preserved and IFN-γ–producing T cells were more suppressed in TNFrsf1b−/− LN-infused recipient mice than in TNFrsf1a−/− LN-infused recipients (supplemental Figure 2), suggesting TNFrsf1b played the larger role in T-cell regulation. Thus, deletion of TNF-αR on donor T cells impaired T-cell infiltration into BM, delayed CD8+ T-cell differentiation to effector memory phenotype, reduced T-cell IFN-γ production, and decreased Fas expression on BM cells to result in overall attenuation of BM destruction. These data suggested active “cross talk” between host-derived TNF-α and donor T cells through TNF-α/TNF-αR interaction.
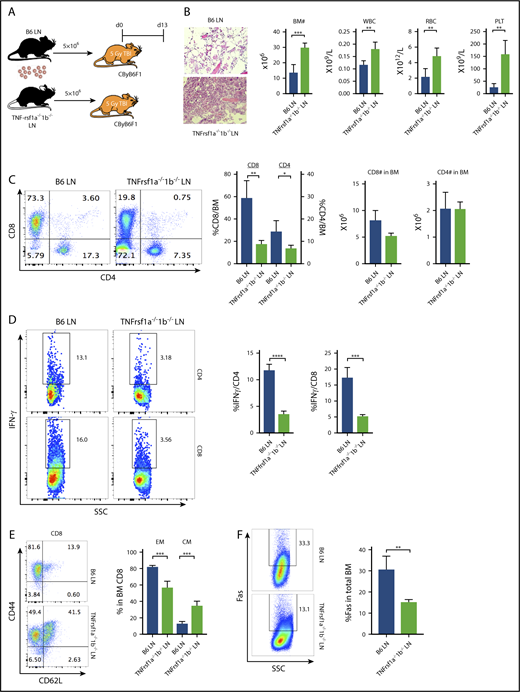
TNF-α modulates T-cell function through engagement with TNF-αRs on T cells. (A) CByB6F1 recipients were preirradiated at 5 Gy TBI and were infused with 5 × 106 LN cells from WT B6 (n = 5) or TNFrsf1a−/−1b−/− (n = 4) donors. Recipient mice were bled and analyzed on day 13. Relative to recipients of WT B6 LN cells, recipients of TNFrsf1a−/−1b−/− LN cells had: (B) higher levels of BM cells, RBCs, WBCs, and platelets (PLT) (original magnification ×200; hematoxylin and eosin stain); (C) lower-level infiltration of T cells, especially CD8+ T cells, in recipient BM; (D) reduced intracellular IFN-γ expression in BM-infiltrated CD4+ and CD8+ T cells; (E) reduced BM proportion of effector memory (EM) CD8+ T cells and increased BM proportion of central memory (CM) CD8+ T cells; (F) reduced Fas expression on residual BM cells. *P < .05; **P < .01; ***P < .001; ****P < .0001.
TNF-α modulates T-cell function through engagement with TNF-αRs on T cells. (A) CByB6F1 recipients were preirradiated at 5 Gy TBI and were infused with 5 × 106 LN cells from WT B6 (n = 5) or TNFrsf1a−/−1b−/− (n = 4) donors. Recipient mice were bled and analyzed on day 13. Relative to recipients of WT B6 LN cells, recipients of TNFrsf1a−/−1b−/− LN cells had: (B) higher levels of BM cells, RBCs, WBCs, and platelets (PLT) (original magnification ×200; hematoxylin and eosin stain); (C) lower-level infiltration of T cells, especially CD8+ T cells, in recipient BM; (D) reduced intracellular IFN-γ expression in BM-infiltrated CD4+ and CD8+ T cells; (E) reduced BM proportion of effector memory (EM) CD8+ T cells and increased BM proportion of central memory (CM) CD8+ T cells; (F) reduced Fas expression on residual BM cells. *P < .05; **P < .01; ***P < .001; ****P < .0001.
To confirm the effect of TNF-α on T-cell IFN-γ secretion and the requirement for TNF-αR on T cells, we compared intracellular IFN-γ levels in B6 WT LN cells and TNFrsf1a−/−1b−/− LN cells in the presence or absence of recombinant TNF-α in vitro. In both CD4+ (Figure 6A) and CD8+ T cells (Figure 6B), addition of TNF-α increased the proportion of IFN-γ–secreting T cells, with less effect in resting cells (Figure 6A-B top panels) and more striking when T cells were activated by PMA plus ionomycin (Figure 6A-B bottom panels). In contrast, addition of TNF-α did not increase levels of intracellular IFN-γ in TNFrsf1a−/−1b−/− CD4+ or CD8+ T cells, either in a resting condition or after stimulation with PMA (Figure 6A-B), suggesting that TNF-α modulated T-cell IFN-γ expression through TNF-αR on T cells. To investigate the molecular changes underlying this effect, we isolated CD8+ T cells from B6 WT or TNFrsf1a−/−1b−/− LNs, stimulated them with PMA plus ionomycin in the presence of TNF-α, and performed PCR array focusing on genes related to T-cell activation and anergy. TNF-α induced higher expression of Ifng (2.3-fold), Tbx21 (6.4-fold), Gzmb (2.3-fold), and other genes in B6 WT CD8+ T cells than did TNFrsf1a−/−1b−/− CD8+ T cells under activation status, but Foxp3 (4.7-fold) and Fasl (three-fold) expression were higher in TNFrsf1a−/−1b−/− CD8+ T cells (Figure 6C). The transcriptome data were consistent with flow cytometry results in revealing higher IFN-γ levels and elevated expression of the master transcriptional regulator Tbx21 and other transcription factors induced by TNF-α.
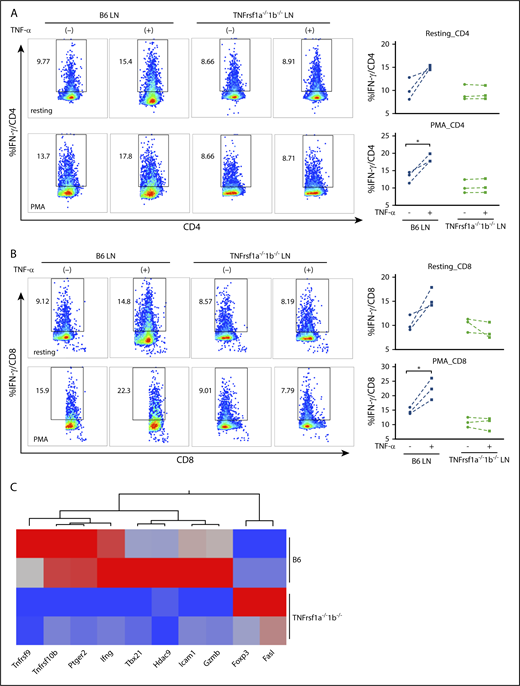
Effects of TNF-α on murine T-cell IFN-γ secretion mediated by TNF-αRs. Recombinant TNF-α (50 ng/mL) were added to LN cells (LN) from WT B6 mice or TNFrsf1a−/−1b−/− mice under resting condition or activated by phorbol 12-myristate-13-acetate (PMA; 5 ng/mL) plus ionomycin (1 μM) overnight. Intracellular IFN-γ levels in CD4+ T cells (A) and CD8+ T cells (B) were examined by flow cytometry. Representative flow cytometry plots from 3 separate experiments are shown. (A-B) Top panels, Under resting condition; bottom panels, after stimulation with PMA plus ionomycin. (C) Transcriptome changes related to the impact of TNF-α on activated T cells. CD8+ T cells from B6 or TNFrsf1a−/−1b−/− mice were stimulated with PMA plus ionomycin in the presence of TNF-α (50 ng/mL) for 4 hours, and cells were then subjected to RNA extraction and complementary DNA (cDNA) synthesis. A PCR array focused on genes related to T-cell activation and anergy was performed. Genes with at least twofold differences between CD8+ T cells from B6 and TNFrsf1a−/−1b−/− mice are shown as a heatmap. Arrays were replicated from 2 different pools of CD8+ T cells. Red indicates high expression; blue, low expression. *P < .05.
Effects of TNF-α on murine T-cell IFN-γ secretion mediated by TNF-αRs. Recombinant TNF-α (50 ng/mL) were added to LN cells (LN) from WT B6 mice or TNFrsf1a−/−1b−/− mice under resting condition or activated by phorbol 12-myristate-13-acetate (PMA; 5 ng/mL) plus ionomycin (1 μM) overnight. Intracellular IFN-γ levels in CD4+ T cells (A) and CD8+ T cells (B) were examined by flow cytometry. Representative flow cytometry plots from 3 separate experiments are shown. (A-B) Top panels, Under resting condition; bottom panels, after stimulation with PMA plus ionomycin. (C) Transcriptome changes related to the impact of TNF-α on activated T cells. CD8+ T cells from B6 or TNFrsf1a−/−1b−/− mice were stimulated with PMA plus ionomycin in the presence of TNF-α (50 ng/mL) for 4 hours, and cells were then subjected to RNA extraction and complementary DNA (cDNA) synthesis. A PCR array focused on genes related to T-cell activation and anergy was performed. Genes with at least twofold differences between CD8+ T cells from B6 and TNFrsf1a−/−1b−/− mice are shown as a heatmap. Arrays were replicated from 2 different pools of CD8+ T cells. Red indicates high expression; blue, low expression. *P < .05.
AA patients have higher frequencies of TNF-α–producing macrophages in BM, and TNF-α can increase IFN-γ expression in human T cells from patients and healthy controls
To assess the relevance of the findings from murine models, we measured macrophages from BM and assessed intracellular TNF-α levels, comparing AA patients with healthy volunteers. In both populations of viable CD3− cells and total BM, we found significantly higher frequencies of CD16+CD68+ cells in AA patients than were present in healthy controls (Figure 7A). CD16+CD68+ cells from either AA patients or healthy controls had increased expression of CD16 and decreased expression of CD14 (Figure 7B), matching the phenotypes of macrophages. Macrophages have higher background staining than T cells due to abundant cell-surface Fc receptors. Mouse immunoglobulin G1, κ isotype control was used to gate TNF-α+ cells in macrophages. The CD16+CD68+ macrophages showed higher levels of intracellular TNF-α than did CD4+ or CD8+ T cells from the same samples (Figure 7C). Greater frequencies of BM macrophages and high intracellular TNF-α in macrophages implicate these cells and this cytokine in the pathophysiology of AA.
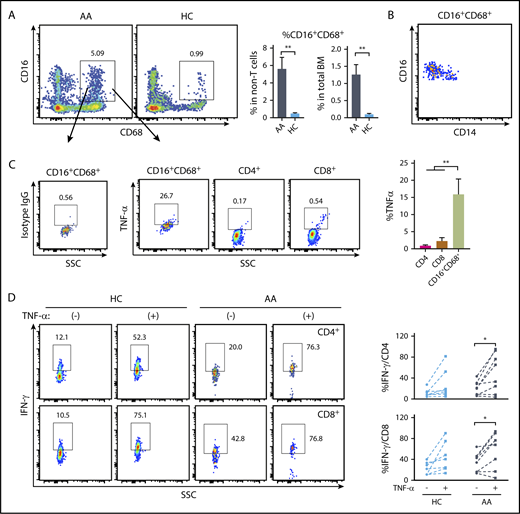
TNF-α expression by BM macrophages from AA patients and impact of TNF-α on human T-cell IFN-γ expression. (A) Comparison of the frequencies of BM macrophages (CD16+CD68+ in viable CD3− population) in AA patients (n = 8) and healthy controls (HC; n = 7). (B) CD14 and CD16 expression in CD16+CD68+ population. (C) Frequencies of TNF-α–producing cells in CD16+CD68+, CD4+, and CD8+ populations in AA patients detected by intracellular staining. Representative dot plots are shown. (D) Peripheral blood mononuclear cells (2 × 106/mL) from AA patients or healthy controls were stimulated with PMA (5 ng/mL) plus ionomycin (1 μM) in the presence or absence of human recombinant TNF-α (100 ng/mL) overnight. Intracellular IFN-γ levels in CD4+ T cells and CD8+ T cells from healthy controls (n = 7) and AA patients (n = 8) were examined by flow cytometry. *P < .05; **P < .01.
TNF-α expression by BM macrophages from AA patients and impact of TNF-α on human T-cell IFN-γ expression. (A) Comparison of the frequencies of BM macrophages (CD16+CD68+ in viable CD3− population) in AA patients (n = 8) and healthy controls (HC; n = 7). (B) CD14 and CD16 expression in CD16+CD68+ population. (C) Frequencies of TNF-α–producing cells in CD16+CD68+, CD4+, and CD8+ populations in AA patients detected by intracellular staining. Representative dot plots are shown. (D) Peripheral blood mononuclear cells (2 × 106/mL) from AA patients or healthy controls were stimulated with PMA (5 ng/mL) plus ionomycin (1 μM) in the presence or absence of human recombinant TNF-α (100 ng/mL) overnight. Intracellular IFN-γ levels in CD4+ T cells and CD8+ T cells from healthy controls (n = 7) and AA patients (n = 8) were examined by flow cytometry. *P < .05; **P < .01.
We also examined IFN-γ secretion by activated human T cells in response to TNF-α. Addition of recombinant TNF-α to PMA and ionomycin-stimulated peripheral blood mononuclear cells enhanced the frequencies of IFN-γ–secreting CD4+ and CD8+ T cells from some, but not all, healthy controls and AA patients (Figure 7D).
Discussion
Consistent with earlier reports,31,32 TNF-α−/− mice carrying germline deletion of the TNF-α gene develop normally and had no gross structural or morphological abnormalities in our study. However, TNF-α−/− mice were resistant to allogenic LN-cell infusion-induced BM destruction in our well-established FVB→B6 LN cell infusion model.24 Previously, it was reported that TNF-α−/− mice were protected from BM destruction relative to WT mice when exposed to sublethal irradiation, suggesting that TNF-α modulates irradiation-induced apoptosis, possibly relevant to BM dysfunction and onset and progression of secondary myelodysplastic syndromes.33 Our data are consistent with these results in confirming a role for TNF-α in BM-cell destruction. The higher level of BM-cell recovery in TNF-α−/− mice relative to controls with or without the infusion of FVB LN cells supports the notion that deletion of TNF-α abrogates irradiation-modulated BM-cell destruction.33
Both IFN-γ and TNF-α have long been postulated as critical inflammatory cytokines in the destruction of BM cells in human AA,2,34 and upregulation of TNF-α has been reported in AA patients.35-37 In our mouse model of immune-mediated BM failure, IFN-γ stimulates Fas upregulation on BM cells to facilitate their destruction by FasL-bearing effector T cells.10 Our current study is aimed at defining the roles of TNF-α in immune-mediated BM failure, which we originally assumed to be similar to IFN-γ. Unexpectedly, we found that T cells lacking TNF-α were fully functional in inducing BM failure, much different from results with IFN-γ−/− mice, the T cells of which were ineffective in inducing BM destruction.10 These initial findings suggested that IFN-γ and TNF-α had different mechanisms of action in murine immune-mediated BM failure.
Deletion of TNF-α in recipient animals prevented BM failure following LN-cell infusion, indicating that host-derived TNF-α was required for the induction of BM destruction. Because TNF-α and IFN-γ were both upregulated in mice with BM failure, we reasoned that host-derived TNF-α exerted BM-destructive effects by modulation of effector T-cell IFN-γ secretion. Cross talk between TNF-α and IFN-γ had been reported earlier. IFN-γ induces TNF-α expression in mouse macrophages by IFN regulatory factor-1 (IRF-1) and IRF-8.38 IFN-γ augments TNF-α expression in human monocytes in response to bacterial endotoxin or lipopolysaccharide by increasing transcription and messenger RNA stability.39 In vitro culture of mouse BM cells with IFN-γ has been shown to upregulate TNF-α messenger RNA expression.10 Conversely, TNF-α promotes CD14+ cell maturation to dendritic cells when they are stimulated with lipopolysaccharides in vitro, which induces naive CD4+ T cells to produce IFN-γ, TNF-α, and IL-17.40 In our work, depletion of TNF-α–producing macrophages in vivo reduced IFN-γ expression by donor CD4+ and CD8+ T cells whereas administration of recombinant TNF-α to TNF-α−/− mice increased IFN-γ expression by BM T cells and enhanced BM loss in the LN-cell infusion model, further supporting a costimulatory regulation loop between TNF-α and IFN-γ in the course of BM destruction.
Biological effects of TNF-α are mediated by 2 structurally similar but functionally distinct receptors, TNFrsf1a and TNFrsf1b.11 The cytoplasmic domains of these 2 TNF-αRs are unrelated, suggesting that they link to different intracellular signaling pathways. Binding of TNF-α to the receptors initiates a complex array of signaling events in response to TNF-αR activation and is responsible for the pleiotropic functional effects of TNF-α,41 mainly through activation of the MAPK stress-signaling cascade including JNK, p38MAPK, and ERK,41-43 as well as the NF-κB transcription factor.43-45 Depletion of TNFrsf1a results in abrogation of TNF signaling, as evidenced by failure of TNF-α to induce NF-κB in genetically deficient T lymphocytes.46 Loss of TNFrsf1a function renders mice resistant to lethal doses of lipopolysaccharides and Staphylococcus aureus enterotoxin B. TNFrsf1a-deficient mice are severely impaired in clearance of bacterial pathogens and readily succumb to infection.47 Thus, TNFrsf1a plays a decisive role in the host’s defense against microorganisms and their pathogenic products.47-49 Expression of TNFrsf1b is far more limited than is expression of TNFrsf1a. TNFrsf1b does not contain a death domain, and its effects include T-cell activation and proliferation via signaling pathways that involve NF-κB, activator protein 1 (AP-1) and MAPKs.50 In our study, depletion of TNF-αRs, including TNFrsf1a−/−, TNFrsf1b−/−, or TNFrsf1a−/−1b−/−, in recipients failed to ameliorate immune-mediated BM failure. The weaker ability of TNF-αR−/− LNs to induce BM failure and the lower capacity of TNF-αR−/− LNs to produce IFN-γ provide the evidence that TNF-α mediates BM destruction through engagement with TNF-αRs on effector T cells, so as to modulate T-cell IFN-γ secretion. Our results correspond with a previous report that TNF signaling through donor T-cell TNFrsf1b, but not TNFrsf1a, is absolutely required for CD8+ effector maturation and for optimal CD4 help for CD8+ T-cell maturation in a murine model of graft-versus-host disease.51 In a separate work, alloreactive T-cell responses were mediated by TNFrsf1a in murine BM transplantation.52 From our results, a plausible sequence in BM failure would be: T cells’ initial activation and secretion of IFN-γ; induction of host macrophages and other cells to produce TNF-α; binding of TNF-α, with positive feedback via TNF-αR; and activation of JNK, p38MAPK, ERK,41-43 and the NF-κB transcription factor43-45 to increase T-cell activation. These transcription factors are also known to bind to the promotors of IFN-γ,53 thus enhancing T-cell IFN-γ expression and further activating T cells. Consistent with this conclusion, our in vitro observation indicated that TNF-α increased T-cell IFN-γ levels only slightly in the resting state and the effect was far more striking when murine T cells were preactivated. Absence of TNF-αR on T cells blocked this response. However, increased IFN-γ secretion by human T cells in response to TNF-α was only observed in some healthy controls and AA patients, likely due to heterogeneity among human subjects and diverse and complex modes of immune and especially macrophage activation. Variability among individuals may also explain paradoxical results in patients receiving anti–TNF-α therapies. Therapeutic activity of TNF biologics in diseases such as rheumatoid arthritis and inflammatory bowel disease might relate directly to inhibition of pathogenic T cells and also blockade of TNF signaling indirectly affecting T-cell activation. For example, in a recent publication, TNF-α stabilized human regulatory T cells via EZH2 and induced immune suppression.54 Blockade of TNF-αRs on murine donor T cells only partially abrogated destruction of hematopoietic cells in our recipient animals, and many other factors likely also participate in modulating the pathogenic immune response and its associated cytokine response.
Our findings may also be relevant to GVHD after organ or BM transplantation. Overproduction of TNF-α by macrophages is associated with GVHD,55,56 but the mechanisms remain to be fully elucidated. Li et al recently reported that depletion of macrophages using clodronate promoted hematopoietic chimerism and increased donor-specific skin allograft tolerance in mice.27 This observation is consistent with TNF-α and IFN-γ reciprocally regulating their production: removal of TNF-α–producing macrophages reduced T-cell IFN-γ secretion and suppressed T-cell–mediated graft rejection to enhance donor engraftment. Results from our macrophage-depletion experiments are concordant with recent published work showing important roles for macrophages in another murine model of marrow failure.57 The higher frequencies of marrow macrophages of AA patients and the relatively abundant TNF-α in macrophages suggest the relevance of animal modeling for discoveries in humans. The interactions of T cells and macrophages and their cytokines in the pathophysiology of marrow destruction in animal models has clear implications for the development of treatments to target macrophages in patients with AA.
We demonstrate in the current study that depletion of TNF-α in recipients, but not in donors, attenuated BM failure. TNF-α exerts its BM-destructive effects not through interaction with TNF-αRs on target cells to cause cell death directly, but indirectly by stimulation of effector T cells to express IFN-γ and to affect T-cell differentiation/activation. These findings may apply to other immune-mediated organ destruction.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgment
This work was supported by the Intramural Research Program of the National Heart, Lung, and Blood Institute, National Institutes of Health.
Authorship
Contribution: X.F. and J.C. conceived, designed, and performed the experiments and analyzed data; X.F. wrote the manuscript; W.S., Z.W., Z.L., and M.H. performed experiments; and N.S.Y. designed experiments, analyzed results, and edited the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Xingmin Feng, Hematology Branch, National Heart, Lung, and Blood Institute, National Institutes of Health, 9000 Rockville Pike, Bethesda, MD 20892; e-mail: fengx2@nhlbi.nih.gov.
