

Key Points
Increased levels of protein C activation cofactors form an anticoagulant vascular domain in CCMs.
Plasma levels of soluble TM may represent a biomarker for CCM risk of hemorrhage.

Abstract
Cerebral cavernous malformations (CCMs) are common brain vascular dysplasias that are prone to acute and chronic hemorrhage with significant clinical sequelae. The pathogenesis of recurrent bleeding in CCM is incompletely understood. Here, we show that central nervous system hemorrhage in CCMs is associated with locally elevated expression of the anticoagulant endothelial receptors thrombomodulin (TM) and endothelial protein C receptor (EPCR). TM levels are increased in human CCM lesions, as well as in the plasma of patients with CCMs. In mice, endothelial-specific genetic inactivation of Krit1 (Krit1ECKO) or Pdcd10 (Pdcd10ECKO), which cause CCM formation, results in increased levels of vascular TM and EPCR, as well as in enhanced generation of activated protein C (APC) on endothelial cells. Increased TM expression is due to upregulation of transcription factors KLF2 and KLF4 consequent to the loss of KRIT1 or PDCD10. Increased TM expression contributes to CCM hemorrhage, because genetic inactivation of 1 or 2 copies of the Thbd gene decreases brain hemorrhage in Pdcd10ECKO mice. Moreover, administration of blocking antibodies against TM and EPCR significantly reduced CCM hemorrhage in Pdcd10ECKO mice. Thus, a local increase in the endothelial cofactors that generate anticoagulant APC can contribute to bleeding in CCMs, and plasma soluble TM may represent a biomarker for hemorrhagic risk in CCMs.
Introduction
Cerebral cavernous malformations (CCMs) are common central nervous system (CNS) vascular anomalies that occur sporadically or heritably and affect ∼1 in 200 humans.1 Mutations of 3 genes, KRIT1 (CCM1), CCM2, or PDCD10 (CCM3), are associated with the development of CCMs, cerebral venous capillary dysplasias with clusters of endothelium filled with blood and prone to hemorrhage.2,3 The KRIT1+/− genotype is the most common cause of the familial form of CCM, whereas the PDCD10+/− genotype often results in a more severe clinical manifestation of the disease.4,5 Unlike sporadic CCM, in which a single lesion is typically observed, familial CCMs are characterized by multiple vascular lesions that are believed to be due to random inactivation of the normal allele.1,4,6 Moreover, a combination of chronic bleeding and episodes of acute hemorrhagic stroke, associated with CCM, causes focal neurological deficits, headaches, epileptic seizures, and, occasionally, death.7 In addition, patients with PDCD10 mutations are more likely to present with significant CCM hemorrhages earlier in life.4 Although the annual symptomatic hemorrhage rate varies largely among studies from 0.25 to 22.9% per patient-year,2,8 it is thought that all CCMs harbor occult bleeding.7,8 Natural history studies and magnetic resonance imaging (MRI) analysis have identified prior CCM bleeding within a year as a predictor of repeated hemorrhage and subsequent clinical sequelae.7,9,10 However, the detailed molecular mechanisms underlying the pathogenesis of CNS hemorrhage in CCM remain elusive.
Recently, we performed genome-wide transcriptome analysis of the acute effects of inactivation of Krit1 in murine brain microvascular endothelial cells (BMECs) and found increased levels of Thbd messenger RNA (mRNA), which encodes the natural anticoagulant receptor thrombomodulin (TM).11 Although TM levels are notably low in normal young brain endothelium, TM plays a role in the thromboresistant properties of the brain.12,13 TM binds thrombin and, while bound, thrombin fails to convert fibrinogen into insoluble fibrin; instead, it catalyzes the formation of activated protein C (APC). APC generation is enhanced by the presence of the endothelial cell protein receptor (EPCR),13,14 the mRNA of which is also increased with loss of Krit1 in BMECs.11 Because APC is a potent natural anticoagulant, and high levels of TM have been associated with a bleeding disorder15,16 and used as a biomarker of endothelial cell dysfunction,17 we hypothesized that increased TM and EPCR could create a local bleeding diathesis in CCM.
Here, we show that TM is upregulated in CCM lesions, as well as in the plasma of individuals with the sporadic or familial form of the disease. In 2 acute models of CCM, we report marked increases in brain endothelial TM and EPCR following genetic inactivation of Krit1 or Pdcd10. Increased expression of TM, but not EPCR, was ascribable to elevation of the transcription factors KLF2 and KLF4 in CCM. Increased TM expression contributes to CCM hemorrhage, because genetic inactivation of 1 or 2 copies of the Thbd gene decreases brain bleeding in Pdcd10ECKO mice. Moreover, administration of blocking antibodies against TM and EPCR reduced CNS bleeding in Pdcd10ECKO mice. Thus, we propose that upregulation of TM and EPCR in CCM endothelium forms an anticoagulant vascular domain that contributes to the bleeding-induced morbidity in CCM.
Material and methods
Patient recruitment
From May of 2015 to November of 2017, 77 patients (mean age, 34.26 ± 18.53 years; range, 5.18-76.02) with a confirmed diagnosis of CCM (38 sporadic, 21 CCM1, and 18 CCM3) and 10 healthy subjects (mean age, 33.26 ± 7.49 years; range, 23.0-43.75) were enrolled in this study. The recruitment of patients was performed in conjunction with their routine clinical evaluations or follow-up. All participants provided informed consent to participate in this research in accordance with the Declaration of Helsinki and according to guidelines approved by The University of Chicago Institutional Review Board. The ethical principles guiding the Institutional Review Board are consistent with The Belmont Report and comply with the rules and regulations of The Federal Policy for the Protection of Human Subjects (56 FR 28003).
As per currently accepted disease categorization, CCM cases were classified as sporadic if they harbored a solitary lesion on the most sensitive susceptibility weighted imaging MRI sequences or a cluster of lesions associated with a developmental venous anomaly.3,10 They were classified as familial if they harbored multifocal CCM lesions, a family history of CCM in a first-degree blood relative, or a mutation genotyped at a CCM gene locus.3,10 Patients with CCM lesion resection or any prior brain irradiation were excluded.
Genetically modified mice
The endothelial-specific conditional Krit1- or Pdcd10-null mice were generated by breeding transgenic mice expressing endothelial-specific Pdgfb promoter–driven tamoxifen-regulated Cre recombinase, iCreERT2,18 with loxP-flanked Krit1 exon 5 (Krit1fl/fl) mice (Pdgfb-iCreERT2; Krit1fl/fl)11 or with loxP-flanked Pdcd10 exon 4 and 5 mice (Pdcd10fl/f; a generous gift from Wang Min, Yale University) (Pdgfb-iCreERT2; Pdcd10fl/fl). All experiments were performed using age-matched Krit1fl/fl or Pdcd10fl/fl littermates on the same C57BL/6 background. Thbdfl/fl mice (a generous gift from Hartmut Weiler, Medical College of Wisconsin) were crossed with Pdgfb-iCreERT2; Pdcd10fl/fl mice to generate Pdgfb-iCreERT2; Pdcd10fl/fl; Thbdfl/wt and Pdgfb-iCreERT2; Pdcd10fl/fl; Thbdfl/fl mice. Mice were administered 50 μg of 4-hydroxy-tamoxifen (Sigma-Aldrich; H7904) by intragastric injection on postnatal day (P)1 to induce Cre activity and endothelial Krit1 or Pdcd10 gene inactivation in the littermates bearing iCreERT2 (Krit1ECKO or Pdcd10ECKO mice). These mice and control Krit1fl/fl or Pdcd10fl/fl mice were euthanized on the indicated days. All animal experiments were carried out in compliance with animal procedure protocols approved by the University of California, San Diego (UCSD) Institutional Animal Care and Use Committee.
Isolation and purification of primary BMECs
Adult mice (2-4 months) were euthanized, and their brains were removed, placed into ice-cold buffer A (10 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid, 1× penicillin-streptomycin, 0.5% bovine serum albumin in Dulbecco’s modified Eagle medium), and processed as previously described.11
Genetic inactivation of Krit1 or Pdcd10 in BMECs
BMECs at passages 1 through 3 were maintained at 37°C in 95% air and 5% CO2, grown to 85% confluence, and treated for 48 hours with 5 μM 4-hydroxy-tamoxifen (Sigma-Aldrich; product number H7904). Thereafter, the medium was replaced with medium lacking 4-hydroxy-tamoxifen,11 and cells were harvested at the time indicated.
Bleeding quantification
Pdcd10ECKO mice and littermate control Pdcd10fl/fl mice were treated with inhibitory antibodies (4.6 mg/kg) against mouse TM (MTM-1701)19 and (2.6 mg/kg) mouse EPCR (rcr-16)20 at P7 and euthanized at P10. Pdcd10ECKO;ThbdECKO/wt, Pdcd10ECKO;ThbdECKO, Pdcd10ECKO, and littermate control Pdcd10fl/fl mice were euthanized at P10. Brains were harvested and transferred into 500 μL of 1× PBS (sample A) and incubated for 3 minutes with gentle agitation. Retinas were transferred into 1 mL of 4% PFA for 20 minutes and incubated at room temperature. After 3 washes with 1× PBS, retinas were dissected to remove cornea and lens and to expose extravascular blood from the retina using 100 μL of 1× PBS (sample B). Then extravascular bleedings from brains (sample A) and retinas (sample B) were processed. For brains, we used 200 μL of sample A, whereas we used 50 μL of sample B plus 150 μL of PBS for retinas. Acetic acid was added to a final concentration of 1% (v/v) to lyse the extravasated red blood cells, and hemoglobin was subsequently measured using a microplate reader (VersaMax; Molecular Devices) set to 405 nm.
Statistical analysis
Data are expressed as mean ± standard error of the mean (SEM). For all experiments, the number of independent experiments (n) is indicated. The sample sizes were estimated with a 2-sample t test (2 tailed). A 2-tailed unpaired Student t test was used to determine statistical significance. For multiple comparisons, 1-way analysis of variance (ANOVA), followed by the Tukey post hoc test, was used.
Data sharing
Supplemental Methods and a table of oligonucleotides used (supplemental Table 1) are available on the Blood Web site.
Results
Increased TM expression in CCM lesions
We recently performed genome-wide transcriptome analysis of the effects of acute inactivation of Krit1 on murine BMECs and observed increased Thbd mRNA that encodes a major anticoagulant endothelial receptor, TM.11 TM expression is modest in normal brain microvasculature relative to systemic blood vessels, suggesting that the brain is a hemostatically “privileged” environment to avert hemorrhage.13,21,22 To examine whether TM expression is increased in human CCM, we stained lesions obtained at surgery. TM staining was increased in the endothelium of human CCM lesions in comparison with lesion-free brain tissue from a CCM patient or nonneurological disease control (Figure 1A). Furthermore, increased expression of TM in CCM endothelial cells was confirmed by an ∼2.9-fold increase in THBD mRNA in CCM lesion endothelial cells isolated by laser capture microdissection (Figure 1B).
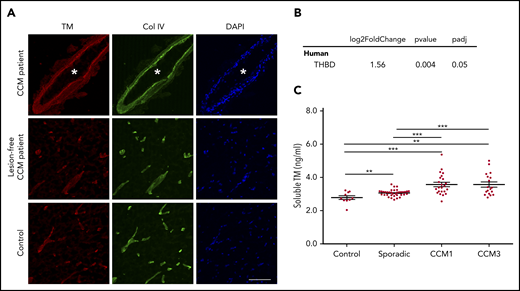
Increased TM in CCM. (A) Immunofluorescence staining of TM (red) and collagen IV (Col IV; green) of human CCM lesion-free brain tissue matched to the CCM patient (2 samples) and brain tissue from a healthy subject (Control). Asterisks denote vascular lumen of CCM lesions. Scale bar, 100 μm (n = 4). (B) Expression levels of THBD, as assessed by RNA sequencing of endothelial cells isolated from human CCM lesions (SEM, n = 5). (C) Plasma levels of soluble TM in patients with sporadic CCM (SEM, n = 38), CCM1 (SEM, n = 21), or CCM3 (SEM, n = 18), as well as normal controls (SEM, n = 10). **P < .01, ***P < .001, unpaired 2-sample t-test.
Increased TM in CCM. (A) Immunofluorescence staining of TM (red) and collagen IV (Col IV; green) of human CCM lesion-free brain tissue matched to the CCM patient (2 samples) and brain tissue from a healthy subject (Control). Asterisks denote vascular lumen of CCM lesions. Scale bar, 100 μm (n = 4). (B) Expression levels of THBD, as assessed by RNA sequencing of endothelial cells isolated from human CCM lesions (SEM, n = 5). (C) Plasma levels of soluble TM in patients with sporadic CCM (SEM, n = 38), CCM1 (SEM, n = 21), or CCM3 (SEM, n = 18), as well as normal controls (SEM, n = 10). **P < .01, ***P < .001, unpaired 2-sample t-test.
We next examined venous plasma levels of soluble TM in familial and sporadic forms of CCM. Significantly increased soluble TM was detected in patients with familial CCM due to mutations in KRIT1 (CCM1) or PDCD10 (CCM3) (3.57 ± 0.64 ng/mL [mean ± SEM]) compared with healthy subjects (2.78 ± 0.11 ng/mL [mean ± SEM]). There was also significantly increased soluble TM in patients with sporadic CCM (3.06 ± 0.03 ng/mL [mean ± SEM]) (Figure 1C). Thus, CCM lesions arising from loss of function of KRIT1 (CCM1) or PDCD10 (CCM3) led to an increase in TM protein and mRNA levels in CCM lesions and an increase in TM in the plasma.
Loss of brain endothelial KRIT1 or PDCD10 increases the expression of TM
To confirm that reduced expression of KRIT1 causes increased TM expression in human brain endothelium, we analyzed the effect of silencing of KRIT1 on TM expression in a human brain endothelial cell line. Infection of hCMEC/D3 cells with a virus encoding short hairpin RNA for KRIT1 (shKRIT1)11 (Figure 2A) significantly increased the expression of TM mRNA and protein (Figure 2B-C). Immunofluorescence also revealed elevated levels of membrane-bound TM protein in KRIT1-depleted human brain endothelial cells (Figure 2D). Flow cytometry analysis showed that surface TM protein increased ∼1.7-fold in KRIT1-depleted cells (Figure 2E-F). An increase in human brain endothelial TM following loss of KRIT1 was accompanied by an increased capacity of brain endothelial cells to generate APC (Figure 2G). The increased APC generation was specific, because blocking antibodies against TM or EPCR reduced APC generation (Figure 2G).
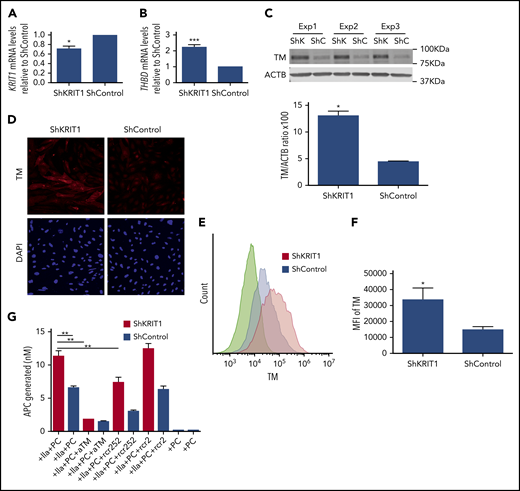
Loss of KRIT1 increases TM in human brain endothelial cells. hCMEC/D3 cells were transduced with short hairpin KRIT1 (shKRIT1) or short hairpin scrambled control (shControl) using lentivirus. (A) shKRIT1 induced an ∼30% decrease in KRIT1 mRNA in hCMEC/D3 cells, as determined by real-time quantitative PCR (RT-qPCR). (B) KRIT1-depleted hCMEC/D3 cells expressed ∼2-fold as much TM mRNA as did ShControl cells (SEM, n = 4). (C) Silencing of KRIT1, using shKRIT1, in hCMEC/D3 cells resulted in an ∼2.9-fold increase in TM protein levels (SEM, n = 3). (D) Representative confocal images of TM (red) staining in hCMEC/D3 cells transduced with shKRIT1 or shControl. Nuclei were counterstained with DAPI (blue). Scale bar, 200 μm (n = 2). (E) Relative abundance of TM in hCMEC/D3 cells transduced with shKRIT1 or shControl, as assessed by flow cytometry. (F) Mean fluorescence intensity (MFI) of membrane-bound TM in shKRIT1 compared with shControl-transduced hCMEC/D3 cells in (B) (SEM, n = 3). (G) Assessment of protein C activation on hCMEC/D3 cells transduced with shKRIT1 or shControl. Thrombin (IIa) and protein C (PC) were added to shKRIT1 or ShControl brain endothelial cells in the presence or absence of blocking antibodies anti-TM (aTM) or anti-EPCR (rcr252). The nonblocking anti-EPCR (rcr2) antibody was used as control (SEM, n = 3). *P < .05, **P < .01, ***P < .001, Student t test.
Loss of KRIT1 increases TM in human brain endothelial cells. hCMEC/D3 cells were transduced with short hairpin KRIT1 (shKRIT1) or short hairpin scrambled control (shControl) using lentivirus. (A) shKRIT1 induced an ∼30% decrease in KRIT1 mRNA in hCMEC/D3 cells, as determined by real-time quantitative PCR (RT-qPCR). (B) KRIT1-depleted hCMEC/D3 cells expressed ∼2-fold as much TM mRNA as did ShControl cells (SEM, n = 4). (C) Silencing of KRIT1, using shKRIT1, in hCMEC/D3 cells resulted in an ∼2.9-fold increase in TM protein levels (SEM, n = 3). (D) Representative confocal images of TM (red) staining in hCMEC/D3 cells transduced with shKRIT1 or shControl. Nuclei were counterstained with DAPI (blue). Scale bar, 200 μm (n = 2). (E) Relative abundance of TM in hCMEC/D3 cells transduced with shKRIT1 or shControl, as assessed by flow cytometry. (F) Mean fluorescence intensity (MFI) of membrane-bound TM in shKRIT1 compared with shControl-transduced hCMEC/D3 cells in (B) (SEM, n = 3). (G) Assessment of protein C activation on hCMEC/D3 cells transduced with shKRIT1 or shControl. Thrombin (IIa) and protein C (PC) were added to shKRIT1 or ShControl brain endothelial cells in the presence or absence of blocking antibodies anti-TM (aTM) or anti-EPCR (rcr252). The nonblocking anti-EPCR (rcr2) antibody was used as control (SEM, n = 3). *P < .05, **P < .01, ***P < .001, Student t test.
We used BMECs isolated from mice bearing floxed alleles of Krit1 (Krit1fl/fl) or Pdcd10 (Pdcd10fl/fl) and an endothelial-specific tamoxifen-regulated Cre recombinase (Pdgfb-iCreERT218 ) to investigate the effect of acute genetic inactivation of CCM genes11 on brain endothelial TM expression. Consistent with results observed in human brain endothelial cells, TM protein levels were significantly increased in 4-hydroxy-tamoxifen–treated Pdgfb-iCreERT2; Krit1fl/fl(Krit1ECKO) BMECs compared with control Krit1fl/fl BMECs (Figure 3A). Increased APC generation was also observed following genetic inactivation of Krit1 in BMECs (data not shown). In addition, acute genetic inactivation of Pdcd10 in 4-hydroxy-tamoxifen–treated Pdgfb-iCreERT2;Pdcd10fl/fl(Pdcd10ECKO) BMECs caused an ∼2.3-fold increase in TM protein levels, as assessed by western blotting (Figure 3B). Consistent with increased protein abundance, RT-qPCR analysis confirmed a dramatic increase in Thbd mRNA levels in Krit1ECKO BMECs (∼8-fold) and Pdcd10ECKO (∼8.8-fold) BMECs compared with control Krit1fl/fl or Pdcd10fl/fl BMECs, respectively (Figure 3C).
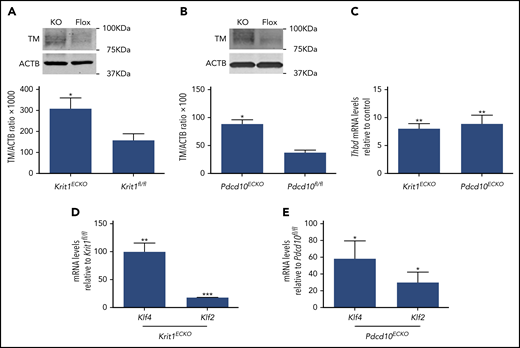
Inactivation of Krit1 or Pdcd10 increases the expression of TM in murine brain endothelial cells. (A) Quantification of TM protein from 3 independent biological replicates in Krit1ECKO (KO) BMECs compared with Krit1fl/fl (Flox) BMECs (SEM, n = 3). (B) Quantification of TM protein from 3 independent biological replicates in Pdcd10ECKO (KO) BMECs compared with Pdcd10fl/fl (Flox) BMECs (SEM, n = 3). (C) RT-qPCR analysis of Thbd mRNA in Krit1ECKO or Pdcd10ECKO BMECs compared with Krit1fl/fl or Pdcd10fl/fl BMECs (control), respectively (SEM, n = 3). Analysis of Klf2 and Klf4 mRNA by RT-qPCR in Krit1ECKO BMECs (D) or in Pdcd10ECKO BMECs (E) compared with Krit1fl/fl or Pdcd10fl/fl BMECs (control), respectively (SEM, n = 3). *P < .05, **P < .01, ***P < .001, Student t test.
Inactivation of Krit1 or Pdcd10 increases the expression of TM in murine brain endothelial cells. (A) Quantification of TM protein from 3 independent biological replicates in Krit1ECKO (KO) BMECs compared with Krit1fl/fl (Flox) BMECs (SEM, n = 3). (B) Quantification of TM protein from 3 independent biological replicates in Pdcd10ECKO (KO) BMECs compared with Pdcd10fl/fl (Flox) BMECs (SEM, n = 3). (C) RT-qPCR analysis of Thbd mRNA in Krit1ECKO or Pdcd10ECKO BMECs compared with Krit1fl/fl or Pdcd10fl/fl BMECs (control), respectively (SEM, n = 3). Analysis of Klf2 and Klf4 mRNA by RT-qPCR in Krit1ECKO BMECs (D) or in Pdcd10ECKO BMECs (E) compared with Krit1fl/fl or Pdcd10fl/fl BMECs (control), respectively (SEM, n = 3). *P < .05, **P < .01, ***P < .001, Student t test.
KLF2 and KLF4 regulate expression of endothelial TM in CCM
Transcription factors KLF2 and KLF4 are regulators of endothelial anticoagulant gene expression.23-26 We confirmed that the changes in brain endothelial TM, at the protein and mRNA levels, were associated with a large increase in transcription factors KLF2 and KLF4 in Krit1ECKO and Pdcd10ECKO BMECs (Figure 3D-E). Therefore, to test whether an increase in KLF2 and KLF4 induces TM during CCM, we used lentivirus-mediated transduction to ectopically express KLF2 and KLF4 in human endothelial cells (Figure 4A) at levels that we previously showed to suppress a target gene, Thbs1.11 Overexpression of KLF2 and KLF4 led to a significant increase in TM mRNA levels compared with control GFP-infected cells (Figure 4B). We also observed that ectopic expression of KLF2 or KLF4 resulted in ∼4.2-fold or ∼5.8-fold increases in TM protein, respectively, as determined by western blot analysis (Figure 4C). Flow cytometry analysis showed that membrane-bound endothelial TM was increased ∼4-fold in KLF4-overexpressing cells compared with control infected cells (Figure 4D-E). Moreover, we observed that reducing the expression of KLF2 and KLF4 (Figure 4F-G) prevented increased TM expression in KRIT1-depleted human brain endothelial cells (Figure 4H). Together, these observations suggest that the increase in KLF2 and KLF4 led to increased brain endothelial TM in CCMs.
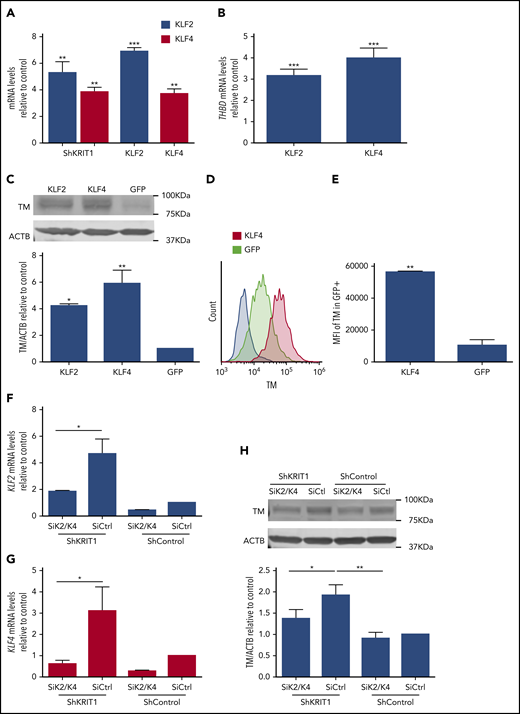
KLF2 and KLF4 regulate expression of endothelial TM. (A) Human umbilical vein endothelial cells (HUVECs) were transduced with lentivirus encoding shKRIT1, KLF2, or KLF4, and the increase in KLF2 or KLF4 mRNA relative to cells transduced with lentivirus control encoding shControl (for shKRIT1) or GFP-Control (for KLF2 or KLF4) was measured by RT-qPCR (SEM, n = 5).35 (B) Analysis of THBD mRNA levels by RT-qPCR in HUVECs transduced with lentivirus encoding KLF2 or KLF4 compared with lentivirus encoding GFP as control (SEM, n = 3 or 4). (C) Analysis of TM protein levels in HUVECs transduced with lentivirus encoding KLF2 or KLF4, as assessed by western blot analysis; lentivirus encoding GFP was used as control (SEM, n = 3). (D) Relative abundance of TM in GFP+ HUVECs transduced with lentivirus encoding KLF4-GFP or GFP as control, as assessed by flow cytometry. (E) Mean fluorescent intensity (MFI) of TM in KLF4-GFP+ cells compared with control GFP+ transduced HUVEC cells in (D) (SEM, n = 3). (F) Analysis of KLF2 mRNA levels by RT-qPCR in hCMEC/D3 cells transduced with lentivirus encoding shKRIT1 or scrambled control, followed by transfection with KLF2 and KLF4–specific small interfering RNAs (siRNAs; siK2/K4) or small interfering RNA control (siCtrl) (SEM, n = 4). (G) Analysis of KLF4 mRNA levels by RT-qPCR in hCMEC/D3 cells transduced with lentivirus encoding shKRIT1 or lentivirus encoding scrambled control, followed by transfection with KLF2 and KLF4–specific siRNAs (siK2/K4) or siRNA control (siCtrl) (SEM, n = 4). (H) Silencing of KLF2 and KLF4 (siK2/K4) using specific siRNAs prevented increased TM expression in KRIT1-depleted hCMEC/D3 cells. Cells were transduced with lentivirus encoding shKRIT1 or with lentivirus encoding shControl (SEM, n = 4). *P < .05, *P < .01, ***P < .001, Student t test.
KLF2 and KLF4 regulate expression of endothelial TM. (A) Human umbilical vein endothelial cells (HUVECs) were transduced with lentivirus encoding shKRIT1, KLF2, or KLF4, and the increase in KLF2 or KLF4 mRNA relative to cells transduced with lentivirus control encoding shControl (for shKRIT1) or GFP-Control (for KLF2 or KLF4) was measured by RT-qPCR (SEM, n = 5).35 (B) Analysis of THBD mRNA levels by RT-qPCR in HUVECs transduced with lentivirus encoding KLF2 or KLF4 compared with lentivirus encoding GFP as control (SEM, n = 3 or 4). (C) Analysis of TM protein levels in HUVECs transduced with lentivirus encoding KLF2 or KLF4, as assessed by western blot analysis; lentivirus encoding GFP was used as control (SEM, n = 3). (D) Relative abundance of TM in GFP+ HUVECs transduced with lentivirus encoding KLF4-GFP or GFP as control, as assessed by flow cytometry. (E) Mean fluorescent intensity (MFI) of TM in KLF4-GFP+ cells compared with control GFP+ transduced HUVEC cells in (D) (SEM, n = 3). (F) Analysis of KLF2 mRNA levels by RT-qPCR in hCMEC/D3 cells transduced with lentivirus encoding shKRIT1 or scrambled control, followed by transfection with KLF2 and KLF4–specific small interfering RNAs (siRNAs; siK2/K4) or small interfering RNA control (siCtrl) (SEM, n = 4). (G) Analysis of KLF4 mRNA levels by RT-qPCR in hCMEC/D3 cells transduced with lentivirus encoding shKRIT1 or lentivirus encoding scrambled control, followed by transfection with KLF2 and KLF4–specific siRNAs (siK2/K4) or siRNA control (siCtrl) (SEM, n = 4). (H) Silencing of KLF2 and KLF4 (siK2/K4) using specific siRNAs prevented increased TM expression in KRIT1-depleted hCMEC/D3 cells. Cells were transduced with lentivirus encoding shKRIT1 or with lentivirus encoding shControl (SEM, n = 4). *P < .05, *P < .01, ***P < .001, Student t test.
An increase in brain endothelial TM is accompanied by hemorrhage in Pdcd10ECKO mice
We used Pdcd10ECKO mice to investigate the distribution and expression of TM protein in experimental murine CCM. At P9, Pdcd10ECKO hindbrains exhibited visible hemorrhagic CCM lesions (Figure 5A); bleeding was confirmed in hematoxylin and eosin–stained sections (Figure 5B) and immunofluorescence staining of sections for erythrocytes using anti-TER119 (Figure 5C). Double staining of P9 Pdcd10ECKO cerebellums for TM and isolectin B4 showed increased TM staining of the luminal surface of CCMs (Figure 5D). In contrast, P9 Pdcd10fl/fl cerebellum showed minimal expression of TM (Figure 5D). To confirm this increase in TM expression, we isolated Pdcd10ECKO hindbrains and quantified TM protein and mRNA levels. Consistent with results observed in vitro and in situ, Pdcd10ECKO hindbrains showed ∼10-fold and ∼3.3-fold increases in TM protein and mRNA levels, respectively, as assessed by western blot or RT-qPCR analysis, respectively (Figure 5E-F). There was no increase in PECAM1 abundance, as assessed by western blot, confirming that the increased TM was not due to increased density of blood vessels (data not shown). Moreover, we observed an increase in TM levels in heart tissue, but not in lung or intestine tissue, from Pdcd10ECKO mice (Figure 5F). We also noted that expression of KLF2 and KLF4 was markedly increased in Pdcd10ECKO hindbrain tissue (Figure 5G), corroborating previous findings.27 Pdcd10ECKO mice developed retinal vascular lesions in the leading front of the vascular plexus with extensive hemorrhages (Figure 5H-I) associated with a dramatic increase in TM staining; the increased bleeding was confirmed by increased hemoglobin (Figure 5I-J). A loss-of-function mutation in KRIT1 is the most common cause of the familial form of CCM. P11 Krit1ECKO hindbrains exhibited an increase in TM staining of the luminal isolectin B4+ CCM endothelial cells (supplemental Figure 1); however, TM staining was clearly less intense than that observed in Pdcd10ECKO CCMs. In contrast with Pdcd10ECKO mice, Krit1ECKO mice exhibited much fewer hemorrhagic CCM lesions (supplemental Figure 1). Collectively, these findings suggested that CCMs in Pdcd10ECKO and Krit1ECKO mice, like humans, express increased levels of TM in lesions that contribute to brain hemorrhage.
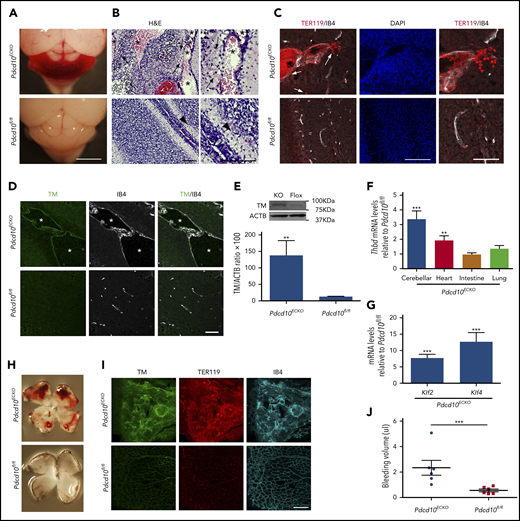
Loss of Pdcd10 increased expression of brain endothelial TM. (A) Pdcd10ECKO hindbrains show prominent hemorrhagic lesions. Scale bar, 2 mm. (B) Hematoxylin and eosin staining of cerebellar sections from Pdcd10ECKO mice and Pdcd10fl/fl littermate controls. Arrows indicate bleeding into the brain parenchyma, and arrowheads indicate normal cerebral vasculature. Asterisks denote the vascular lumen of CCM lesions. Higher-magnification images are shown (right panels). Scale bars, 100 μm (n = 4 mice in each group). (C) Immunofluorescence staining of blood cells using TER119 (red) and labeling of the microvasculature, using isolectin B4 (white), of cerebellar sections from Pdcd10ECKO or Pdcd10fl/fl mice at P9; DAPI staining (blue) was used to reveal nuclei (left and middle panels). Arrows indicate bleeding into the brain parenchyma. Scale bar, 100 μm. Higher-magnification images are shown (right panels; scale bar, 50 μm). (n = 3 mice in each group). (D) Immunofluorescence staining of TM (green) or isolectin B4 (IB4; white) of cerebellar sections from Pdcd10ECKO and Pdcd10fl/fl mice. Asterisks denote the vascular lumen of CCM lesions. Scale bar, 50 μm (n = 4 mice in each group). (E) Quantification of TM in cerebellar tissue in Pdcd10ECKO and control Pdcd10fl/fl littermates, as assessed by western blot analysis (SEM, n = 4 or 7 mice in each group). (F) Analysis of Thbd mRNA levels by RT-qPCR in cerebellar, heart, intestine, and lung tissue from Pdcd10ECKO mice compared with control Pdcd10fl/fl littermates (SEM, n = 6 or 8 mice in each group). (G) Analysis of Klf2 and Klf4 mRNA levels by RT-qPCR in cerebellar tissue in Pdcd10ECKO mice compared with control Pdcd10fl/fl littermates (SEM, n = 7 mice in each group). (H) Whole-mount P9 retinas show hemorrhage in Pdcd10ECKO mice but not in littermate control Pdcd10fl/fl mice. (I) Maximum-intensity projection of whole-mount P9 retinal vasculature at the angiogenic growth front stained for TM (green) and for red blood cells using TER119 antibodies (red); retinal vasculature was labelled with isolectin B4 (IB4; turquoise) (n = 4). (J) Measurement of hemoglobin content of extravasated erythrocytes from retinal vasculature at P10 in Pdcd10ECKO mice and control Pdcd10fl/fl littermates. **P < .01, ***P < .001, Student t test and 1-way ANOVA, followed by the Tukey post hoc test.
Loss of Pdcd10 increased expression of brain endothelial TM. (A) Pdcd10ECKO hindbrains show prominent hemorrhagic lesions. Scale bar, 2 mm. (B) Hematoxylin and eosin staining of cerebellar sections from Pdcd10ECKO mice and Pdcd10fl/fl littermate controls. Arrows indicate bleeding into the brain parenchyma, and arrowheads indicate normal cerebral vasculature. Asterisks denote the vascular lumen of CCM lesions. Higher-magnification images are shown (right panels). Scale bars, 100 μm (n = 4 mice in each group). (C) Immunofluorescence staining of blood cells using TER119 (red) and labeling of the microvasculature, using isolectin B4 (white), of cerebellar sections from Pdcd10ECKO or Pdcd10fl/fl mice at P9; DAPI staining (blue) was used to reveal nuclei (left and middle panels). Arrows indicate bleeding into the brain parenchyma. Scale bar, 100 μm. Higher-magnification images are shown (right panels; scale bar, 50 μm). (n = 3 mice in each group). (D) Immunofluorescence staining of TM (green) or isolectin B4 (IB4; white) of cerebellar sections from Pdcd10ECKO and Pdcd10fl/fl mice. Asterisks denote the vascular lumen of CCM lesions. Scale bar, 50 μm (n = 4 mice in each group). (E) Quantification of TM in cerebellar tissue in Pdcd10ECKO and control Pdcd10fl/fl littermates, as assessed by western blot analysis (SEM, n = 4 or 7 mice in each group). (F) Analysis of Thbd mRNA levels by RT-qPCR in cerebellar, heart, intestine, and lung tissue from Pdcd10ECKO mice compared with control Pdcd10fl/fl littermates (SEM, n = 6 or 8 mice in each group). (G) Analysis of Klf2 and Klf4 mRNA levels by RT-qPCR in cerebellar tissue in Pdcd10ECKO mice compared with control Pdcd10fl/fl littermates (SEM, n = 7 mice in each group). (H) Whole-mount P9 retinas show hemorrhage in Pdcd10ECKO mice but not in littermate control Pdcd10fl/fl mice. (I) Maximum-intensity projection of whole-mount P9 retinal vasculature at the angiogenic growth front stained for TM (green) and for red blood cells using TER119 antibodies (red); retinal vasculature was labelled with isolectin B4 (IB4; turquoise) (n = 4). (J) Measurement of hemoglobin content of extravasated erythrocytes from retinal vasculature at P10 in Pdcd10ECKO mice and control Pdcd10fl/fl littermates. **P < .01, ***P < .001, Student t test and 1-way ANOVA, followed by the Tukey post hoc test.
EPCR expression in the brain is upregulated in CCM but is not regulated by KLF2 or KLF4
EPCR, an endothelial transmembrane protein that presents protein C to the thrombin–TM complex14 is expressed in the brain vasculature and plays a crucial role in the vascular anticoagulant pathway28,29 by facilitating protein C activation.30 We next investigated whether EPCR levels were also altered in CCMs. PROCR (EPCR) mRNA and protein expression was increased in human CCMs in comparison with control brain tissue (Figure 6A-B). RT-qPCR and western blot analysis confirmed the upregulation of EPCR mRNA and protein in Krit1ECKO and Pdcd10ECKO BMECs (Figure 6C-E). Consistent with results observed in vitro, Pdcd10ECKO hindbrains showed an ∼2-fold increase in Procr (Epcr) mRNA abundance (Figure 6F). Increased Procr mRNA was specific to lesion areas, because we did not observe an increase in other vascular beds of Pdcd10ECKO mice analyzed (Figure 6F). In contrast to TM, ectopic expression of KLF2 or KLF4 did not change EPCR protein or mRNA levels in endothelial cells, as assessed by western blot or RT-qPCR analysis, respectively (Figure 6G-H).
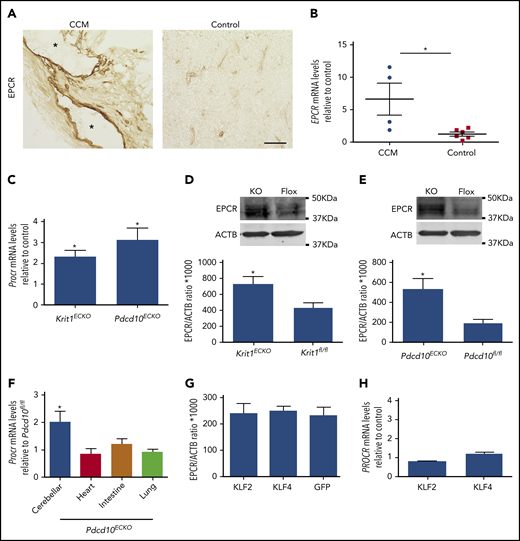
Increased EPCR in CCMs. (A) Immunohistochemistry of EPCR (brown precipitate) of human CCMs and brain tissue from a healthy subject as control. Asterisks denote vascular lumen of CCM lesions. Scale bar, 100 μm (n = 3). (B) Analysis of PROCR (EPCR) mRNA levels by RT-qPCR in human CCM lesions or control brain tissue in (A) (SEM, n = 4 CCM tissue, n = 1 lesion-free brain tissue from CCM patient, n = 2 healthy subject brain tissue, and n = 2 postmortem specimens as control). (C) RT-qPCR analysis of Procr (Epcr) mRNA in Krit1ECKO or Pdcd10ECKO BMECs compared with Krit1fl/fl or Pdcd10fl/fl BMECs (control), respectively (SEM, n = 3). (D) Quantification of EPCR protein in 9-day Krit1ECKO (KO) BMECs compared with Krit1fl/fl (Flox) BMECs (SEM, n = 3). (E) Quantification of EPCR protein in 20-day Pdcd10ECKO (KO) BMECs compared with Pdcd10fl/fl (Flox) BMECs (SEM, n = 3). (F) Analysis of Epcr mRNA levels by RT-qPCR in cerebellar, heart, intestine, and lung tissue in Pdcd10ECKO mice and control Pdcd10fl/fl littermates (SEM, n = 4 or 7 mice in each group). (G) Analysis of EPCR protein levels in human umbilical vein endothelial cells transduced with lentivirus encoding KLF2 or KLF4, as assessed by western blot analysis; lentivirus encoding GFP was used as control (SEM, n = 3). (H) Analysis of EPCR mRNA levels by RT-qPCR in human umbilical vein endothelial cells transduced with lentivirus encoding KLF2 or KLF4 compared with lentivirus encoding GFP as control (SEM, n = 3 or 4). *P < .05, Student t test and 1-way ANOVA, followed by the Tukey post hoc test.
Increased EPCR in CCMs. (A) Immunohistochemistry of EPCR (brown precipitate) of human CCMs and brain tissue from a healthy subject as control. Asterisks denote vascular lumen of CCM lesions. Scale bar, 100 μm (n = 3). (B) Analysis of PROCR (EPCR) mRNA levels by RT-qPCR in human CCM lesions or control brain tissue in (A) (SEM, n = 4 CCM tissue, n = 1 lesion-free brain tissue from CCM patient, n = 2 healthy subject brain tissue, and n = 2 postmortem specimens as control). (C) RT-qPCR analysis of Procr (Epcr) mRNA in Krit1ECKO or Pdcd10ECKO BMECs compared with Krit1fl/fl or Pdcd10fl/fl BMECs (control), respectively (SEM, n = 3). (D) Quantification of EPCR protein in 9-day Krit1ECKO (KO) BMECs compared with Krit1fl/fl (Flox) BMECs (SEM, n = 3). (E) Quantification of EPCR protein in 20-day Pdcd10ECKO (KO) BMECs compared with Pdcd10fl/fl (Flox) BMECs (SEM, n = 3). (F) Analysis of Epcr mRNA levels by RT-qPCR in cerebellar, heart, intestine, and lung tissue in Pdcd10ECKO mice and control Pdcd10fl/fl littermates (SEM, n = 4 or 7 mice in each group). (G) Analysis of EPCR protein levels in human umbilical vein endothelial cells transduced with lentivirus encoding KLF2 or KLF4, as assessed by western blot analysis; lentivirus encoding GFP was used as control (SEM, n = 3). (H) Analysis of EPCR mRNA levels by RT-qPCR in human umbilical vein endothelial cells transduced with lentivirus encoding KLF2 or KLF4 compared with lentivirus encoding GFP as control (SEM, n = 3 or 4). *P < .05, Student t test and 1-way ANOVA, followed by the Tukey post hoc test.
Blocking TM and EPCR attenuates vascular hemorrhage in Pdcd10ECKO mice
To assess the role of endogenous endothelial TM in CCM hemorrhages, we examined the impact of genetic inactivation of endothelial Thbd in Pdcd10ECKO mice. Pdcd10ECKO;ThbdECKO/wt mice exhibited a significant reduction in brain bleeding (∼1.6-fold decrease), as did Pdcd10ECKO;ThbdECKO mice (∼1.8-fold decrease), compared with Pdcd10ECKO littermates (Figure 7A). Moreover, the upregulation of TM and EPCR contributed to brain hemorrhage in Pdcd10ECKO mice; blocking antibodies against mouse TM and mouse EPCR reduced bleeding, as signaled by an ∼2-fold reduction in extractable hemoglobin in Pdcd10ECKO mice (Figure 7B). These data suggest that CCMs caused by mutations of KRIT1 or PDCD10 lead to the formation of an anticoagulant vascular domain due to increased expression of TM and EPCR that is driven, in part, by transcription factors KLF2 and KLF4, thereby contributing to increased hemorrhage (Figure 7C).
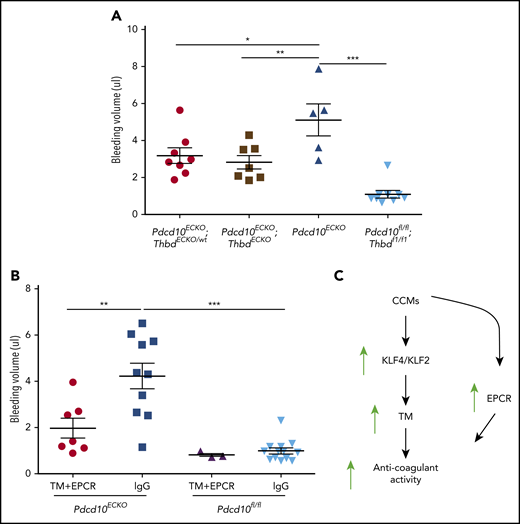
TM and EPCR create an anticoagulant vascular domain in CCMs. Measurement of hemoglobin content of extravasated erythrocytes from hindbrains at P10. (A) Decreased cerebellar hemorrhages are observed in Pdcd10ECKO;ThbdECKO/wt or Pdcd10ECKO;ThbdECKO mice compared with Pdcd10ECKO littermates. Pdcd10fl/fl is a non-CCM lesion control (SEM, n = 5 or 8). (B) Blocking antibodies against TM (4.6 μg/g) and EPCR (2.6 μg/g) or immunoglobulin G (7.2 μg/g) control were injected retro-orbitally into Pdcd10ECKO mice or control Pdcd10fl/fl littermate controls at P7 (SEM, n = 3 or 13). (C) Schematic diagram of the formation of an anticoagulant domain in CCM lesion. Increased expression of KLF2 and KLF4 transcription factors lead to upregulation of brain endothelial TM. In addition, EPCR expression is upregulated in CCMs independently of KLF2 and KLF4. Increased levels of TM and EPCR create anticoagulant domains that predispose the CCM lesions to hemorrhages.
TM and EPCR create an anticoagulant vascular domain in CCMs. Measurement of hemoglobin content of extravasated erythrocytes from hindbrains at P10. (A) Decreased cerebellar hemorrhages are observed in Pdcd10ECKO;ThbdECKO/wt or Pdcd10ECKO;ThbdECKO mice compared with Pdcd10ECKO littermates. Pdcd10fl/fl is a non-CCM lesion control (SEM, n = 5 or 8). (B) Blocking antibodies against TM (4.6 μg/g) and EPCR (2.6 μg/g) or immunoglobulin G (7.2 μg/g) control were injected retro-orbitally into Pdcd10ECKO mice or control Pdcd10fl/fl littermate controls at P7 (SEM, n = 3 or 13). (C) Schematic diagram of the formation of an anticoagulant domain in CCM lesion. Increased expression of KLF2 and KLF4 transcription factors lead to upregulation of brain endothelial TM. In addition, EPCR expression is upregulated in CCMs independently of KLF2 and KLF4. Increased levels of TM and EPCR create anticoagulant domains that predispose the CCM lesions to hemorrhages.
Discussion
CNS bleeding is a primary source of morbidity and mortality in CCM patients. We report that CCM endothelial cells manifest elevated expression of TM and EPCR mRNA and protein. This cell-autonomous increased expression of TM and EPCR is a direct result of a decrease in the products of KRIT1 or PCDC10, 2 genes whose loss of function leads to CCM formation. An increase in the expression of KLF2 and KLF4, 2 transcription factors already implicated in CCM pathogenesis, can account for increased TM expression; however, these transcription factors did not account for the upregulation of EPCR expression. The increased abundance of TM and EPCR on the endothelial cell surface results in an increased capacity to support the generation of APC, a potent endogenous anticoagulant that inactivates the coagulation cofactors factor Va and factor VIIa.22 Increased TM expression levels contributed to CCM hemorrhage, because genetic inactivation of 1 or 2 copies of the Thbd gene in endothelial cells decreases brain bleeding in Pdcd10ECKO mice. Moreover, administration of blocking antibodies against TM and EPCR significantly reduced CCM hemorrhage in Pdcd10ECKO mice. These studies identify that an increase in expression of 2 endothelial cofactors generates anticoagulant APC in CCM and indicate that the formation of an anticoagulant endothelial domain in CCM lesions can contribute to bleeding. Thus, manipulation of the balance between anticoagulant and cytoprotective effects of APC could serve to reduce the incidence and morbidity of CCM bleeding.
The elevation in endothelial surface TM and EPCR can contribute to bleeding in CCM. Cerebral capillary dysplasias with hemosiderin deposition detected by MRI10,31 or histology3,10,32 are hallmarks of CCM lesions. Electron microscopic studies have documented loss of tight junctions in some CCMs,33 and KRIT1 enables Rap1 stabilization of endothelial cell–cell junctions.34,35 Indeed, we have recently found that loss of tight junctions is 1 of the earliest manifestation of inactivation of Krit1 or Pcdc10 in BMECs.11 Disruption of brain interendothelial junctions in CCMs11,36,37 could trigger the recurring microhemorrhages that cause hemosiderin deposition. Importantly, bleeding is much less evident in other cerebrovascular diseases that exhibit loss of blood–brain barrier integrity due to disruption in brain endothelial tight and adherens junctions.38 Normal brain vasculature maintains restricted levels of TM and EPCR compared with systemic blood vessels,21,39 suggesting that the brain is a prohemostatic environment. Our finding that loss of CCM1 or CCM3 causes a marked increase in EPCR and TM on the surface of CCM endothelial cells suggests that CCMs form an anticoagulant vascular domain within the brain that causes a local bleeding diathesis.
Familial and sporadic forms of CCMs exhibited significant increases in soluble TM in plasma, suggesting that this may be a biomarker for patients who are at elevated risk for bleeding. Elevated levels of TM in plasma have been documented and are associated with endothelial dysfunction17,40 and bleeding disorders.15-17 As discussed above, the increased TM can reflect an increased formation of APC in lesions which, in turn, leads to anticoagulant activity13 that facilitates bleeding. As the number of potentially new therapeutic interventions in CCMs grows,11,41-44 the identification of markers to identify patients with an elevated risk for hemorrhage becomes imperative. Indeed, recent studies45,46 showed that levels of inflammatory and angiogenic molecules in peripheral blood plasma could predict CCM clinical behavior, and this study now identifies soluble TM as a new putative biomarker candidate in this disease.
Sangwung et al47 showed that endothelial cell loss of KLF2 and KLF4 results in substantial loss of TM, and we report here that overexpression of these transcription factors can drive TM expression, as would be predicted from previous studies.23-25 There is now compelling evidence that elevation of KLF2 and KLF4 makes important contributions to the pathogenesis of CCM.11,27,41,48 Endothelial-specific deletion of KLF2 and KLF4 in adults leads to lethality,47 indicating that inhibition of these transcription factors is unlikely to be a suitable therapeutic strategy. Here, we add upregulation of TM to the growing list11 of KLF2 and KLF4 targets that can contribute to the pathogenesis of CCM disease and might serve as therapeutic targets. In contrast, we could not ascribe the increase in EPCR expression to KLF2 and KLF4. In particular, depleting KRIT1 increased EPCR and TM expression in cultured endothelial cells, whereas overexpression of KLF2 and KLF4 only increased TM. Thus, the cell autonomous increase in EPCR does not appear to be a consequence of increased expression of KLF2 and KLF4 or of TM. The links between loss of CCM genes and increased EPCR expression could be a fruitful area for future work.
This study suggests new potential avenues for the management of CNS hemorrhage in CCM. Insights gleaned from bleeding in hemophilia increased our awareness that excessive or unbalanced APC generation can actively contribute to bleeding and that the anticoagulant activity of APC is a valid target for drug development.49,50 However, APC is a multifunctional protease and, independently of its anticoagulant activity, APC induces cytoprotective effects on endothelial cells and neurons that are dependent on protease-activated receptor 1 and 3, as well as on EPCR. These cell-signaling activities of APC are central to the primary mechanism of action for APC’s neuroprotective effects in a variety of acute and chronic neuropathologies, including ischemic stroke, traumatic brain injury, chronic cerebral ischemia, amyotrophic lateral sclerosis, and multiple sclerosis.51 Therefore, strategies targeting the protein C system and aimed at mitigating or preventing bleeding in CCMs need to be specific for APC anticoagulant activity while leaving APC’s neuroprotective activities unaltered. This was also illustrated by our experiments in which inhibition of all APC activities (anticoagulant and cytoprotective), using the anti-mouse APC antibody SPC-54,50 caused premature death and cerebral thrombosis in Pdcd10ECKO mice (data not shown). Several anticoagulant-specific strategies targeting the protein C pathway have been proposed for control of bleeding in hemophilia patients with inhibitors, including anti-APC and anti-protein S antibodies specifically targeting APC’s anticoagulant activity and APC-resistant factor Va.49,50 Whether these strategies can be safely adapted to CCM will have to be determined, because the safety window in hemophilia and CCM is likely quite different; targeting of acute CNS bleeds vs the risk of rebleeding in the 1-year window of increased vulnerability that follows such bleeds will be an important consideration for this.7,8
Another consideration is that the high level of APC in CCM lesions could diminish morbidity by means of its cytoprotective effects, which include protection of vascular and blood–brain barrier integrity.14,28 Indeed, signaling-selective 3K3A-APC with minimal anticoagulant activity reduced tissue-type plasminogen activator–induced bleeding in a murine ischemic stroke model,52 indicating that the consequences of enhanced APC generation in CCM might be more complex. New studies are needed to appreciate these complexities and to evaluate which strategies targeting the different components of the protein C system will be most advantageous in CCM. This study motivates the development of such novel strategies for the management of CCM hemorrhage and suggests new biomarkers that could serve to identify those patients with an elevated risk for bleeding as potential subjects for such new therapeutic strategies.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Angioma Alliance for providing human CCM specimens; Selina Chang, Riya Verma, Tiffany S. Shamas, and Shady Soliman for technical assistance; Jennifer Santini for microscopy technical assistance; and the UCSD School of Medicine Microscopy Core. The anti-EPCR antibodies (rcr-252, rcr-2, and rcr-16) were generously made available by Kenji Fukudome (Saga Medical School, Saga, Japan). Thbdfl/fl mice were the generous gift of Hartmut Weiler (Blood Research Institute, Milwaukee, WI).
This work was supported by National Institutes of Health, National Institute of Neurological Disorders and Stroke grant NS092521 (M.H.G. and I.A.A.) and National Institutes of Health, National Heart, Lung, and Blood institute grants K01 HL133530 (M.A.L.-R.), HL104165, HL130678, and HL142975 (L.O.M.), and HL078784 (M.H.G.), as well as by the UCSD School of Medicine Microscopy Core (P30 NS047101).
Authorship
Contribution: M.A.L.-R. designed and performed the experiments, analyzed and interpreted the data, generated the figures, and wrote the manuscript; A.P. and P.H. performed and analyzed histological and gene expression experiments in culture and conditional knockout mice; R.G., J.K., L.S., S.P., and I.A.A. performed, analyzed, and interpreted the RNA transcriptome of human CCM lesions and human TM plasma levels; T.W., A.Y., and L.O.M. performed, analyzed, and interpreted protein C activation and bleeding studies; R.G., A.P., P.H., T.W., A.Y., I.A.A., and L.O.M. helped to write the manuscript; F.L., I.A.R., and L.O.M. provided reagents and helped to interpret the data; C.T.E. provided critical reagents; and M.H.G. conceived the project, designed the overall study, analyzed and interpreted data, and wrote the manuscript.
Conflict-of-interest disclosure: UCSD and The Scripps Research Institute hold intellectual property rights related to APC-resistant factor Va and some uses of APC mutants for which L.O.M. is listed as inventor. L.O.M. is a founder and a member of the Board of Directors of Hematherix LLC. The remaining authors declare no competing financial interests.
Correspondence: Mark H. Ginsberg, Department of Medicine, Leichtag Biomedical Research Building, University of California San Diego, La Jolla, CA 92093; e-mail: mhginsberg@ucsd.edu.
