

Key Points
Leukemia blast-derived superoxide induces a senescent phenotype in cells within the BM microenvironment.
Targeting the senescent BM cells improves the survival of mice with leukemia.

Abstract
Acute myeloid leukemia (AML) is an age-related disease that is highly dependent on the bone marrow (BM) microenvironment. With increasing age, tissues accumulate senescent cells, characterized by an irreversible arrest of cell proliferation and the secretion of a set of proinflammatory cytokines, chemokines, and growth factors, collectively known as the senescence-associated secretory phenotype (SASP). Here, we report that AML blasts induce a senescent phenotype in the stromal cells within the BM microenvironment and that the BM stromal cell senescence is driven by p16INK4a expression. The p16INK4a-expressing senescent stromal cells then feed back to promote AML blast survival and proliferation via the SASP. Importantly, selective elimination of p16INK4a+ senescent BM stromal cells in vivo improved the survival of mice with leukemia. Next, we find that the leukemia-driven senescent tumor microenvironment is caused by AML-induced NOX2-derived superoxide. Finally, using the p16-3MR mouse model, we show that by targeting NOX2 we reduced BM stromal cell senescence and consequently reduced AML proliferation. Together, these data identify leukemia-generated NOX2-derived superoxide as a driver of protumoral p16INK4a-dependent senescence in BM stromal cells. Our findings reveal the importance of a senescent microenvironment for the pathophysiology of leukemia. These data now open the door to investigate drugs that specifically target the “benign” senescent cells that surround and support AML.
Introduction
Acute myeloid leukemia (AML) is an age-related, often lethal disease that is highly dependent on the bone marrow (BM) microenvironment.1 Despite remission often seen after chemotherapy, long-term survival is modest. Thus, improved outcomes may depend on novel treatment strategies derived from a better understanding of the role of the BM in AML progression and relapse.
The BM is a structurally complex organ comping blood vessels and a heterogeneous population of cells that either directly participate in the generation of blood cells or support the hematopoietic function of the tissue.2 Support cells in the BM all contribute to stimuli required for regulating normal hematopoiesis. In leukemia, however, the BM fails to produce adequate numbers of mature blood cells and platelets.2 Notably, AML blasts have been show to alter the function of BM stromal cells (BMSCs; including endothelial cells, fibroblasts, and adipocytes).1,3-5 This blast cell nonautonomous activity ultimately reshapes the BM microenvironment, thereby promoting AML tumor cell survival and proliferation.
Cellular senescence was described >5 decades ago by Hayflick et al6 as an irreversible arrest of normal cell proliferation. It is now clear that the senescence growth arrest evolved, at least in part, to suppress the development of cancer.7 In addition to arrested growth, senescent cells show widespread changes in chromatin organization and gene expression.8 These changes include the secretion of numerous proinflammatory cytokines, chemokines, growth factors, and proteases, a feature termed the senescence-associated secretory phenotype (SASP).9 The senescence response, therefore, likely evolved not only to suppress the development of cancer, but also to aid tissue repair and regeneration in response to injury. However, because senescent cells gradually increase with age, the senescence response likely becomes maladaptive with age, and there is mounting evidence that they contribute to a variety of age-related phenotypes and pathologies.7,10 Furthermore, the SASP can disrupt a number of cellular and tissue functions, including, ironically, distinct pathologic protumoral changes.7,11
AML is primarily a disease of the elderly, with peak incidence between the ages of 80 and 85 years.12 Because AML is an age-related disease and highly dependent on the BM microenvironment, which naturally becomes senescent with age, we hypothesized, and subsequently tested the idea, that AML not only favors a senescent microenvironment but might actively shape a senescent microenvironment to promote tumor proliferation and survival.
Materials and methods
Materials
Anti-p16 antibody was from BD Biosciences (Oxford, UK). Anti-humanCD45-BV421, anti-mouse CD45-human-BV421, anti-mouse CD31-PerCP, anti-mouse Ter119-antigen-presenting cell (APC), CD33-APC, anti-mouse CD45-PerCP anti-mouse CD140a APC-Cy7, and anti-mouse CD105-fluorescein isothiocyanate were from Miltenyi Biotec (Bergisch Gladbach, Germany) and BioLegend (London, UK). All other reagents were obtained from Sigma-Aldrich (St Louis, MO), unless otherwise indicated.
Methods
Cell cultures
Primary AML blasts and BMSC were obtained from patient BM (supplemental Table 1, available on the Blood Web site). Nonsenescent BMSC were collected from the BM beneath the exposed acetabular surface of the pelvis during surgery of patients undergoing elective hip replacement. CD34+ cells were enriched from whole cord blood using CD34+ Magnetic Bead separation (Miltenyi Biotec). All tissue was taken following informed consent and under approval from the UK National Health Service Health Research Authority (LRECref07/H0310/146). For primary cell isolation, heparinized blood was isolated by density centrifugation using Histopaque as previously described.13 Primary cells and cell lines were cultured in growth medium comprising Dulbecco’s modified Eagle medium (DMEM), 10% fetal bovine serum, and 1% l-glutamine at 5% CO2 at 37°C. BMSC were isolated from hip replacement patients and AML bone marrow samples by adherence to tissue culture plastic and then expanded in DMEM containing 20% fetal bovine serum plus 1% penicillin-streptomycin. BMSC markers were confirmed by flow cytometry for expression of CD90+, CD73+, CD105+, and CD45−.
Coculture experiments
BMSC were seeded at 1 × 104 cells per well of a 4-well plate in normal growth media. BMSC were then transduced with either lentivirus containing control short hairpin (shRNA) or test shRNA (p16 or p21) for 5 days. AML blasts (0.25 × 106) or normal CD34+ cells (0.25 × 106) were then cocultured with the BMSC. Cells counts were based on hemocytometer counting; we used flow cytometry as a second test to confirm the cells were either AML (CD45+/CD33+) or CD34 (CD34+) of origin.
Senescence β-galactosidase assay
Senescence β-galactosidase staining kit (Cell Signaling Technology, MA) was used to detect senescence-associated β-galactosidase (SA-βgal) in BSMC. BMSC were cultured and treated in 35-mm dishes. The BMSC were evaluated for the blue color, indicative of SA-βgal+ cells.
Flow cytometry analysis and cell sorting
Cells were collected by centrifugation for 3 minutes at 400g, washed, and resuspended in phosphate-buffered saline (PBS), incubated for 5 minutes with the Fc receptor blocking reagent and then stained with anti-human CD45-fluorescein isothiocyanate, anti-mouse CD45-PerCP, and anti-mouse CD105-APC antibodies or isotype controls. Analysis were performed on a CytoFLEX and using CytExpert analysis software (Beckman Coulter). Cell sorting was performed on a BD FACSMelody and a BD Aria II cell sorter (both BD Bioscience).
RNA extraction and real-time PCR
Total RNA was extracted from cells using the ReliaPrep RNA extraction kit from Promega (Southampton, UK) according to the manufacturer’s instructions. Reverse transcription was performed using the qPCRBIO complementary DNA synthesis kit (PCR Biosystems, London, UK). Relative quantitative real-time polymerase chain reaction (PCR) using qPCRBIO SyGreen Mix (PCR Biosystems) was performed on complementary DNA generated from purified RNA. After preamplification (95°C for 2 minutes), the PCRs were amplified for 45 cycles (95°C for 15 seconds, 60°C for 10 seconds, and 72°C for 10 seconds) on a 384-well LightCycler 480 (Roche, Burgess Hill, UK). Each messenger RNA (mRNA) level was normalized against the β-actin mRNA level.
Western immunoblotting and enzyme-linked immunosorbent assays
Western analyses following sodium dodecyl sulfate-polyacrylamide gel electrophoresis were carried out as described previously.14 Briefly, whole cell lysates were extracted and sodium dodecyl sulfate-polyacrylamide gel electrophoresis gel electrophoresis separation was performed. Western analysis was performed using anti-p16INKa antibody; membranes were reprobed for β-actin as a loading control.
shRNA silencing of p16, p21, and NOX2
Mission shRNA-targeted lentivirus plasmids were purchased from Sigma-Aldrich. Control and targeted lentivirus stocks were produced as described,15 concentrated using Amicon Ultra centrifugal filters and titers determined using the Lenti-X quantitative reverse transcription PCR titration kit (ClonTech, Saint-Germain-en-Laye, France). BMSC were infected with p16INK4a or p21 targeted virus. Knockdown was confirmed 5 days later and BMSC were then combined with AML cells at a ratio of 1:5. MN1 cells were infected with NOX2-targeted virus. shRNA transduced cells were not sorted or enriched before use in experiments.
Retroviral AML transplantation mouse model
All animal experiments were performed in accordance with UK Home Office approvals and regulations and with approval from the Animal Welfare and Ethical Review Board of the University of East Anglia. C57BL/6J mice were obtained from Charles River (UK). In this study, we also used p16-3MR mice developed by the Campisi laboratory,16 which carry a transgene composed of the p16INK4a regulatory elements driving expression of a fusion protein containing Renilla luciferase (to visualize p16INK4a expressing cells), monomeric red fluorescent protein (RFP; to visualize and isolate p16INK4a expressing cells) and the herpes simplex virus thymidine kinase (to selectively kill such cells by administering the prodrug ganciclovir [GCV])16
BM was harvested from mice, and lineage-negative (lin−) cells were obtained by negative selection using the Lineage Cell Depletion Kit (Miltenyi Biotec). Lin− cells from C57BL/6J were retrovirally infected as previously described.1 To generate MN1 and HoxA9/Meis1 BM cells were harvested from mice, and lin− cells were obtained by negative selection using the Lineage Cell Depletion Kit (mouse) as recommended by the manufacturer (Miltenyi Biotec). Lin− cells derived from C57BL/6J were retrovirally infected by coculture with GP+E86 cells (ATCC) transfected with pSF91MN1iGFP, or MIG-HA-HoxA9 or MIY-HA-Meis1a (kindly provided by Professor Keith Humphries, Terry Fox Laboratory, Vancouver, Canada) in the presence of polybrene (10 µg/mL, Sigma-Aldrich).17 Coculture with GP+E86 packaging pSF91MN1iGFP, MIG-HA-HoxA9 was performed for 3 days. MIH-HA-HoxA9 transfected cells were then cocultured GP+E86 packaging MIY-HA-Meis1a to generate HoxA9/Meis1 and MN1 overexpressing lin− cells. Cells were sorted using an FACS BD Aria II cell sorter. Cells were maintained in DMEM, 20% fetal calf serum supplemented with murine interleukin-3 (IL-3; 10 ng/mL), murine IL-6 (10 ng/mL), and murine SCF (100 ng/mL) all purchased from PeproTech (London, UK). MN1 cells were then infected with pCDH-luciferase-T2A-mCherry, kindly provided by Professor Irmela Jeremias, (Helmholtz Zentrum München, Munich, Germany).18 MN1 cells were plated at 5 × 104/well in 12-well plates and expanded. Transduced MN1 cells (MN1-luc) were sorted using mCherry fluorescence on a FACSAria II cell sorter.
For AML-induced senescence, 8- to 12-week old p16-3MR mice were injected with 1 × 105 MN1-luc expressing cells. To determine engraftment of MN1-luc cells into p16-3MR and C57BL/6 animals, mice were injected intraperitoneally (IP) with luciferin (Fisher Scientific) and imaged for firefly luminescence (MN1-luc) using the In-vivo Bruker Xtreme (Coventry, UK). For survival experiments, MN1-engrafted p16-3MR mice were split into 2 groups before treatment with GCV. Mice were then injected IP with GCV (25 mg/kg) or PBS (control) for 5 consecutive days.
Primary AML and CD34+ hematopoietic progenitor cell xenograft model
NOD.Cg-Prkdcscid IL2rgtm1Wjl/SzJ (NSG) mice were obtained from The Jackson Laboratory (Bar Harbor, ME). NSG mice were maintained under specific pathogen-free conditions. For the AML xenograft model, 2 × 106 viable primary AML cells were washed and resuspended in PBS. Cells were then injected into the tail vein of nonirradiated 6- to 8-week-old female NSG mice. When clinical signs of illness were apparent (rough fur, hunchback, or reduced motility) or if 12 weeks postinjection was reached, mice were euthanized by exposure to CO2. For the CD34+ hematopoietic progenitor cell (HPC) xenograft model (humanized NSG [hu-NSG]) 2 × 105 CD34+ cord blood cells were injected into the tail vein of nonirradiated 3- to 4-week-old female NSG mice. CD34+ cell engraftment was monitored peripheral blood analysis for human CD45 cells. Twelve weeks postinjection, mice were euthanized by exposure to CO2. BM was harvested and cell sorting was performed for human CD45− and mouse CD45−, Ter119−, CD31−, CD105+, and CD140a+. BM was also analyzed for human cell engraftment by human CD45+ cells. If >1% of human CD45 cells were detected in the BM, the AML sample was determined to be engrafted.
Statistical analyses
The Mann-Whitney U test was used to compare test groups where stated. P < .05 was considered statistically significant and is denoted by an asterisk. Results were the mean ± standard deviation of ≥4 independent experiments. We generated statistics using GraphPad Prism5 software (GraphPad, San Diego, CA). For western analyses, data are representative images of 3 independent experiments.
Results
AML induces a senescent phenotype in vivo
We showed that primary human AML cells induce SASP (upregulation of IL-6, IL-8, MIP1a, and MIP3a) when cocultured with primary BMSC.4 Here, we determined whether AML cells can induce SASP in vivo. We engrafted 4 primary AML patient-derived cells (AML-NSG) and normal human cord blood–derived CD34+ HPC (hu-NSG) into immunocompromised NSG mice by tail vein injection (schematic, Figure 1A). Ten to 12 weeks after injection, we euthanized the animals; we determined engraftment (Figure 1B), sorting CD45− (mouse and human), CD31−, Ter119−, CD140a+, and CD105+ BMSC (Figure 1C). The frequency of BMSC in hu-NSG compared with AML-NSG is shown in supplemental Figure 1. Mouse BMSC (mBMSC) from hu-NSG served as a negative control. Sorted BMSC from hu-NSG and AML-NSG were analyzed for gene expression by real-time PCR. Our analyses showed that the expression of p16INK4a, and to a lesser extent p21, was consistently upregulated in mBMSC from AML cell–engrafted mice (Figure 1D). Further, the SASP components IL-6 and MIP3, although not MIP1a, were also consistently upregulated in mBMSC from animals engrafted with leukemic cells compared with animals engrafted with human CD34+ HPC (Figure 1E). Loss of lamin B1 is another identifier of senescent cells.19 Consistent with the expression of p16INK4a, IL-6, and MIP1a, lamin B expression declined in BMSC from AML-engrafted NSG mice (Figure 1F). Plasma concentrations of mouse IL-6 are also elevated in AML-NSG mice compared with hu-NSG mice (Figure 1G). Mouse MIP3 concentrations were undetected in both treatment groups. Together, these data suggest that AML cells can induce a senescent phenotype in the BM microenvironment.
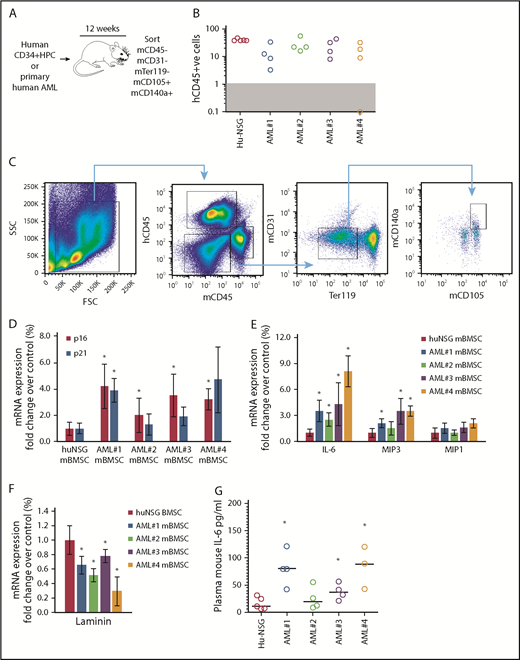
Primary human AML induces a p16-driven SASP in vivo. (A) Schematic of in vivo experiment in which 2 × 106 primary AML cells (4 individual patient AML cells and 5 CD34 HPC) were injected into NSG mice. (B) Engraftment was measured by detecting human CD45 by flow cytometry. In the dot plot, each AML engraftment into NSG mice is shown for BM. (C) Representative gating strategy for BMSC cell (hCD45−, mCD45−, mCD31−, mTer119−, mCD105+, and mCD140a+) population that was sorted. (D) RNA analysis for p16 and p21 in the sorted BMSC (hCD45−, mCD45−, mCD31−, mTer119−, mCD105+, and mCD140a+) isolated from NSG mice engrafted with primary human AML or cord blood CD34+ HPC. (E) RNA analysis for SASP in the sorted BMSC. (F) RNA analysis for lamin B in the sorted BMSC. (G) Terminal peripheral blood samples were taken and plasma isolated from all NSG mice engrafted with primary human AML or cord blood CD34+ HPC and mouse IL-6 was measured by enzyme-linked immunosorbent assay. The Mann-Whitney U test was used to compare between treatment groups (*P < .05). FSC, forward scatter; SSC, side scatter.
Primary human AML induces a p16-driven SASP in vivo. (A) Schematic of in vivo experiment in which 2 × 106 primary AML cells (4 individual patient AML cells and 5 CD34 HPC) were injected into NSG mice. (B) Engraftment was measured by detecting human CD45 by flow cytometry. In the dot plot, each AML engraftment into NSG mice is shown for BM. (C) Representative gating strategy for BMSC cell (hCD45−, mCD45−, mCD31−, mTer119−, mCD105+, and mCD140a+) population that was sorted. (D) RNA analysis for p16 and p21 in the sorted BMSC (hCD45−, mCD45−, mCD31−, mTer119−, mCD105+, and mCD140a+) isolated from NSG mice engrafted with primary human AML or cord blood CD34+ HPC. (E) RNA analysis for SASP in the sorted BMSC. (F) RNA analysis for lamin B in the sorted BMSC. (G) Terminal peripheral blood samples were taken and plasma isolated from all NSG mice engrafted with primary human AML or cord blood CD34+ HPC and mouse IL-6 was measured by enzyme-linked immunosorbent assay. The Mann-Whitney U test was used to compare between treatment groups (*P < .05). FSC, forward scatter; SSC, side scatter.
BMSC senescence is driven by p16INK4a-expressing cells and is protumoral
To determine the drivers of AML-induced senescence in human BMSC, we cultured primary AML cells or normal CD34+ HPC with human primary nonsenescent BMSC for 6 days. Human BMSC were then assayed for senescence-associated SA-βgal, IL-6, and lamin B1. Human BMSC cocultured with AML cells showed increased SA-βgal staining (Figure 2A-B), increased IL-6 and IL-8 expression (Figure 2C), and reduced lamin B1 expression (not shown) compared with BMSC cocultured with normal CD34+ HPC, consistent with AML-induced cellular senescence. Coculture with AML cells also induced p16INK4a and p21 expression in BMSC (Figure 2D). Subsequently, we examined the effect of p16INK4a or p21 knockdown (supplemental Figure 2A-C) in human BMSC on AML cell or normal CD34+ HPC survival. AML cells showed reduced growth, as determined by cell number, when cocultured on p16 knockdown BMSC (Figure 2E; supplemental Figure 2C). In addition, knockdown of p16INK4a in BMSC prevented senescence when cultured with AML (Figure 2F). We conclude that p16 driven senescence in human BMSCs is protumoral.
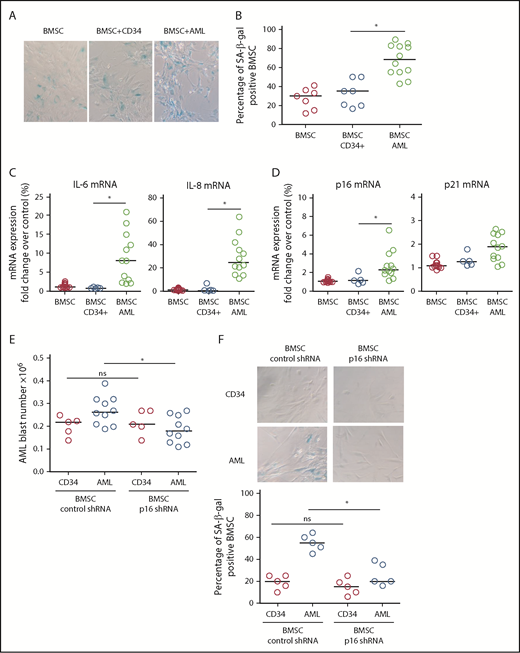
AML-induced senescence in BMSC. (A) BMSC were cultured alone or with primary AML (0.25 × 106; n = 12) or with CD34+ HPC (0.25 × 106; n = 7) for 6 days. Nonadherent cells were removed and BMSC were analyzed for SA-βgal. (B) Bar graph representation of SA-βgal+ cells from panel A. (C) BMSC were cultured alone or with primary AML (0.25 × 106; n = 10) or with CD34+ HPC (0.25 × 106; n = 5) for 6 days. Nonadherent cells were removed and RNA was extracted from the BMSC. RNA was analyzed for IL-6 and IL-8 expression using quantitative reverse transcription PCR. (D) As for panel C, but analyzed for p16 and p21. (E) BMSC were infected with p16 targeted shRNA or control shRNA lentivirus and cultured for 5 days. AML blasts (0.25 × 106; n = 10) or CD34+ HPC (0.25 × 106; n = 5) were cocultured with BMSC with control shRNA or on BMSC with p16 shRNA. AML blast number was assessed using a trypan blue exclusion hemocytometer-based count and CD33/CD45+ staining using flow cytometry. (F) To confirm the senescent profile of BMSC from (E) nonadherent, cells were removed and BMSC were analyzed for senescence associated SA-βgal (n = 5). The Mann-Whitney U test was used to compare between treatment groups (*P < .05). Each dot on the dot plots represents a different AML or CD34 HPC sample. ns, not significant.
AML-induced senescence in BMSC. (A) BMSC were cultured alone or with primary AML (0.25 × 106; n = 12) or with CD34+ HPC (0.25 × 106; n = 7) for 6 days. Nonadherent cells were removed and BMSC were analyzed for SA-βgal. (B) Bar graph representation of SA-βgal+ cells from panel A. (C) BMSC were cultured alone or with primary AML (0.25 × 106; n = 10) or with CD34+ HPC (0.25 × 106; n = 5) for 6 days. Nonadherent cells were removed and RNA was extracted from the BMSC. RNA was analyzed for IL-6 and IL-8 expression using quantitative reverse transcription PCR. (D) As for panel C, but analyzed for p16 and p21. (E) BMSC were infected with p16 targeted shRNA or control shRNA lentivirus and cultured for 5 days. AML blasts (0.25 × 106; n = 10) or CD34+ HPC (0.25 × 106; n = 5) were cocultured with BMSC with control shRNA or on BMSC with p16 shRNA. AML blast number was assessed using a trypan blue exclusion hemocytometer-based count and CD33/CD45+ staining using flow cytometry. (F) To confirm the senescent profile of BMSC from (E) nonadherent, cells were removed and BMSC were analyzed for senescence associated SA-βgal (n = 5). The Mann-Whitney U test was used to compare between treatment groups (*P < .05). Each dot on the dot plots represents a different AML or CD34 HPC sample. ns, not significant.
Deleting BM senescent cells reduces tumor growth
A number of murine models have been developed to study p16INK4a-driven senescence in vivo.16,20,21 We used p16-3MR mice, which carry a transgene comprising the p16INK4a regulatory elements driving expression of a fusion protein containing Renilla luciferase (to visualize p16INK4a-expressing cells), RFP (to isolate such cells) and the herpes simplex virus thymidine kinase (to selectively kill such cells by administering the prodrug GCV) (Figure 3A).16
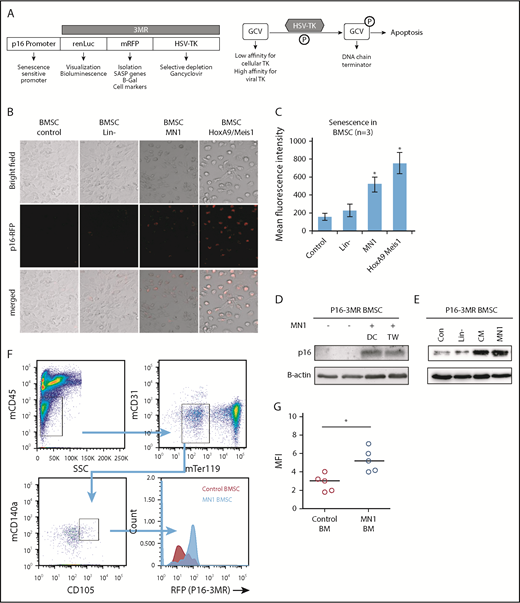
MN1 engraftment drives p16-3MR. (A) Schematic of p16-3MR model. (B) Fluorescent images of p16-3MR–isolated BMSC that have been cultured alone or with lin−, MN1, or HoxA9/Meis1 cells for 6 days (n = 3). (C) Flow cytometry analysis of p16-3MR BMSC that have been cultured alone or with lin−, MN1, or HoxA9/Meis1 cells for 6 days (n = 3). (D) Western blot analysis of p16-3MR BMSC cultured alone or with MN1 for 6 days. Blots were reprobed with B-actin to confirm sample loading (shown are representative images of 3 blots). (E) Western blot analysis of p16-3MR BMSC cultured alone or with lin−, CM from MN1 cells, or MN1 cells. Blots were reprobed with B-actin to confirm sample loading (shown are representative images of 3 blots). (F) 1 × 105 MN1 cells were injected into the tail vein of p16-3MR mice. BM was isolated and analyzed for mouse BMSC (mCD45−, mCD31−, mTer119−, mCD105+, and mCD140a+) expressing RFP using flow cytometry (n = 5). (G) Flow cytometry analysis of p16-3MR BMSC measuring RFP (F). The Mann-Whitney U test was used to compare between treatment groups (*P < .05). CM, conditioned media; DC, direct contact; HSV-TK, herpes simplex virus-1 thymidine kinase; TW, transwell.
MN1 engraftment drives p16-3MR. (A) Schematic of p16-3MR model. (B) Fluorescent images of p16-3MR–isolated BMSC that have been cultured alone or with lin−, MN1, or HoxA9/Meis1 cells for 6 days (n = 3). (C) Flow cytometry analysis of p16-3MR BMSC that have been cultured alone or with lin−, MN1, or HoxA9/Meis1 cells for 6 days (n = 3). (D) Western blot analysis of p16-3MR BMSC cultured alone or with MN1 for 6 days. Blots were reprobed with B-actin to confirm sample loading (shown are representative images of 3 blots). (E) Western blot analysis of p16-3MR BMSC cultured alone or with lin−, CM from MN1 cells, or MN1 cells. Blots were reprobed with B-actin to confirm sample loading (shown are representative images of 3 blots). (F) 1 × 105 MN1 cells were injected into the tail vein of p16-3MR mice. BM was isolated and analyzed for mouse BMSC (mCD45−, mCD31−, mTer119−, mCD105+, and mCD140a+) expressing RFP using flow cytometry (n = 5). (G) Flow cytometry analysis of p16-3MR BMSC measuring RFP (F). The Mann-Whitney U test was used to compare between treatment groups (*P < .05). CM, conditioned media; DC, direct contact; HSV-TK, herpes simplex virus-1 thymidine kinase; TW, transwell.
To determine whether AML induces senescence in p16-3MR mice, we isolated BMSC from these mice and cocultured them with MN1 cells (mouse myeloid leukemic cells), HoxA9/Meis1 (mouse myeloid leukemic cells), or lin− for 6 days; this incubation time has been shown to increase RFP in cells from the p16-3MR model under injury.16 After 6 days of coculture, p16-3MR BMSC cultured with MN1 or HoxA9/Meis1 cells but not in− cells had detectable red fluorescence (Figure 3B-C), consistent with p16INK4a-driven senescence. To confirm senescence in the p16-3MR BMSC, we examined p16INK4a protein levels and showed that, under coculture conditions, p16-3MR BMSC had elevated levels of p16INK4a protein, and this increase did not depend on direct cell–cell contact (Figure 3D). Moreover, conditioned media from MN1 also increased p16INK4a protein expression in BMSC, whereas coculture of BMSC with in− cells had no effect on p16INK4a protein expression (Figure 3E). This finding suggests that the AML signal that drives BMSC into senescence is soluble.
To determine whether AML engraftment induces senescence in the p16-3MR BM in vivo, first we transfected MN1 cells with firefly luciferase (MN1-luc) to detect their presence after engraftment into p16-3MR mice (supplemental Figure 3). Next, firefly luciferase activity confirmed engraftment of MN1 into p16-3MR mice. Finally, we examined BMSC from the BM of p16-3MR animals engrafted with MN1 for RFP (Figure 3F-G). MN1 cells induced RFP expression in p16-3MR mice compared with control animals, consistent with the AML cells inducing a senescence response.
Next, we created senescent p16-3MR BMSC by culturing them with MN1 cells in transwells for 6 days. We then treated the cocultures with GCV (Figure 4A). MN1 cells showed reduced cell number when cultured on senescent BMSC treated with GCV compared with BMSC treated with PBS. To test the hypothesis that senescent BMSC fuel tumorigenesis in vivo, we injected MN1 cells into p16-3MR mice, followed by GCV treatment (Figure 4B). p16-3MR mice engrafted with MN1-luc cells for 14 days, then treated with GCV (once daily for 5 days), showed reduced tumor load compared with PBS-treated p16-3MR mice engrafted with MN1-luc cells (Figure 4C-D). Tumor load was detected by firefly luciferase expressed from MN1-luc cells, which is activated by the substrate luciferin, and not coelenterazine, which activates luciferase derived from Renilla (supplemental Figure 3). Moreover, MN1-luc–engrafted p16-3MR mice survived longer following 5 days of GCV treatment compared with control-treated animals (Figure 4E). Finally, to confirm that this survival was due to the GCV effect of the p16-3MR transgene and not a direct effect of GCV, we engrafted MN1-luc cells into nontransgenic C57BL/6 animals (Figure 4F). GCV had no effect on C57BL/6 mice engrafted with MN1-luc cells. Furthermore, GCV reduced RFP expressing BSMC cells in vivo in p16-3MR mice engrafted with MN1 cells and reduced β-gal staining in BMSC cultured with MN1 cells in vitro (supplemental Figure 4A-B). We conclude that deletion of senescent nonmalignant BMSC is antileukemic.
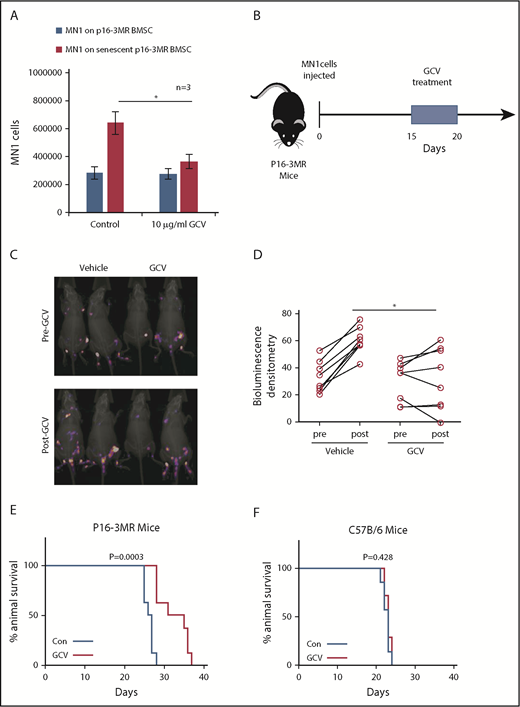
Deleting senescent cells reduces AML tumor volume. (A) MN1 were grown on p16-3MR nonsenescent or senescent BMSC for 2 days with and without treatment with GCV (10 µg/mL) (n = 3). (B) GCV experiment in vivo. (C-D) 1 × 105 MN1-luc cells were injected into p16-3MR mice (n = 8 for each treatment group). Mice were imaged at 14 days postengraftment. At day 15, GCV (25 mg/kg) or PBS treatment was started for 5 days. Mice were then imaged again 1 day after GCV treatment had finished. (C) Pre and post images show the same mice in the same order. (D) Densitometry of the bioluminescent images was performed to determine differences between vehicle and GCV treated animals. (E-F) Kaplan-Meier survival curves for p16-3MR (n = 8) and C57BL/6 (n = 7) mice injected with MN1 and then treated with vehicle or GCV as shown in panel B.
Deleting senescent cells reduces AML tumor volume. (A) MN1 were grown on p16-3MR nonsenescent or senescent BMSC for 2 days with and without treatment with GCV (10 µg/mL) (n = 3). (B) GCV experiment in vivo. (C-D) 1 × 105 MN1-luc cells were injected into p16-3MR mice (n = 8 for each treatment group). Mice were imaged at 14 days postengraftment. At day 15, GCV (25 mg/kg) or PBS treatment was started for 5 days. Mice were then imaged again 1 day after GCV treatment had finished. (C) Pre and post images show the same mice in the same order. (D) Densitometry of the bioluminescent images was performed to determine differences between vehicle and GCV treated animals. (E-F) Kaplan-Meier survival curves for p16-3MR (n = 8) and C57BL/6 (n = 7) mice injected with MN1 and then treated with vehicle or GCV as shown in panel B.
NOX2-derived superoxide drives AML-associated BM senescence
By what mechanism do AML cells induce senescence in BMSC? One established inducer of senescence is oxidative stress,22,23 and we showed that human AML cells induce oxidative stress in BMSC.24 Indeed, MN1 and HoxA9/Meis1 cells, but not lin− cells, increased oxidative stress as measured by increased dihydrodichlorofluorescein diacetate (DCF, detect cellular reactive oxygen species [ROS] level) fluorescence in BMSC from p16-3MR mice (Figure 5A). Supplemental Figure 5 shows that human primary AML, but not normal CD34+ HPC, can induce ROS in BMSC. AML blasts were reported to generate more superoxide than nonmalignant CD34+ cells.25 This ROS was a consequence of superoxide generated by NOX2, which was shown to benefit malignant cells.24,25 Moreover, ROS in the form of H2O2 can induce senescence in several cell types.22,23 Therefore, we determined whether H2O2-induced senescence in BMSCs in culture. Indeed, treating human BMSC with H2O2 induced a senescent phenotype (Figure 5B-C). Importantly, after MN1 cells were injected into allogeneic mice, hydrogen peroxide levels as measured by the Amplex Red Assay were elevated in the whole BM, a phenomenon that did not occur in control mice (Figure 5D). Moreover, ROS levels, analyzed by flow cytometry, in BMSC also increased in MN1 engrafted mice (Figure 5E).
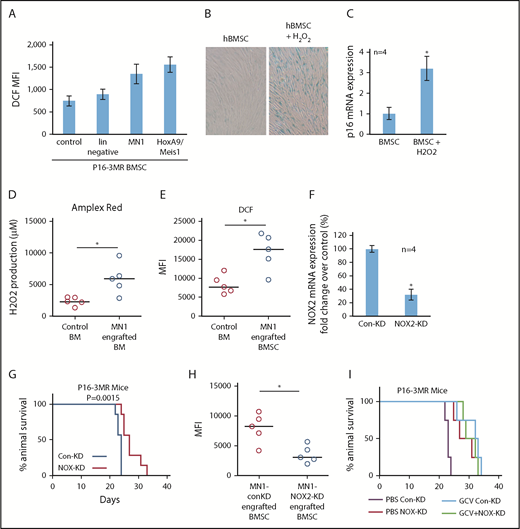
AML-induced NOX2-derived superoxide drives BM senescence. (A) BMSC from p16-3MR were isolated and then cultured alone or with lin−, MN1, or HoxA9/Meis1 cells for 3 days. DCF fluorescence was assessed in BMSC by flow cytometry (n = 4). (B) Human BMSC were treated with 10 μM H2O2 for 6 days and then analyzed for senescence associated SA-βgal, and (C) p16 mRNA expression (n = 4). (D) C57BL/6 mice injected with MN1. At 21 days postengraftment, mice were euthanized and whole BM was isolated and analyzed for H2O2 production using the Amplex red assay (n = 5). (E) C57BL/6 mice were injected with MN1 cells. At 21 days postengraftment, mice were euthanized and BMSC were analyzed by flow cytometry for DCF fluorescence (n = 5). (F) Real-time PCR assay was used to analyze the NOX2 mRNA expression level in NOX2-KD MN1 cells compared with control-KD cells (n = 4). (G) Kaplan-Meier survival curves for p16-3MR mice injected with MN1 NOX2-KD cells or MN1 control-KD cells (n = 7 in each group). (H) At the end point of the experiment, BM was isolated and flow cytometry was performed to detect BMSC-derived RFP (n = 5). (I) Kaplan-Meier survival curves for p16-3MR mice injected with MN1 NOX2-KD cells or MN1 control-KD cells and then injected IP with PBS or GCV at day 15, GCV (25 mg/kg) for 5 days (n = 4).
AML-induced NOX2-derived superoxide drives BM senescence. (A) BMSC from p16-3MR were isolated and then cultured alone or with lin−, MN1, or HoxA9/Meis1 cells for 3 days. DCF fluorescence was assessed in BMSC by flow cytometry (n = 4). (B) Human BMSC were treated with 10 μM H2O2 for 6 days and then analyzed for senescence associated SA-βgal, and (C) p16 mRNA expression (n = 4). (D) C57BL/6 mice injected with MN1. At 21 days postengraftment, mice were euthanized and whole BM was isolated and analyzed for H2O2 production using the Amplex red assay (n = 5). (E) C57BL/6 mice were injected with MN1 cells. At 21 days postengraftment, mice were euthanized and BMSC were analyzed by flow cytometry for DCF fluorescence (n = 5). (F) Real-time PCR assay was used to analyze the NOX2 mRNA expression level in NOX2-KD MN1 cells compared with control-KD cells (n = 4). (G) Kaplan-Meier survival curves for p16-3MR mice injected with MN1 NOX2-KD cells or MN1 control-KD cells (n = 7 in each group). (H) At the end point of the experiment, BM was isolated and flow cytometry was performed to detect BMSC-derived RFP (n = 5). (I) Kaplan-Meier survival curves for p16-3MR mice injected with MN1 NOX2-KD cells or MN1 control-KD cells and then injected IP with PBS or GCV at day 15, GCV (25 mg/kg) for 5 days (n = 4).
Finally, we asked whether AML derived NOX2-dependent superoxide is responsible for inducing p16INK4a-driven senescence in p16-3MR mice. We depleted NOX2 from MN1 cells (Figure 5F). Strikingly, we found increased survival of p16-3MR mice engrafted with MN1 cells with NOX2 knockdown compared with control knockdown animals (Figure 5G). Moreover, BMSC cultured with NOX-KD cells had reduced levels of ROS compared with control-KD MN1 cells (supplemental Figure 6A). Knockdown of NOX2 in MN1 cells engrafted into p16-3MR mice also resulted in significantly fewer senescent BMSC (Figure 5H; supplemental Figure 6B). Moreover, when we treated NOX2-KD MN1 engrafted p16-3MR mice with GCV, no additive affect was observed (Figure 5I). Together, these data identify AML generated NOX2-derived superoxide as a driver of p16INK4a-dependent senescence.
Discussion
Here we report that AML cells induce SASP in the BM microenvironment, which supports the survival and proliferation of the leukemic blasts. In vivo, AML cells induce senescence in BMSC. Deletion of these senescent p16INK4a-expressing BMSC slows tumor progression and extends animal survival. Notably, the senescence response and SASP is driven by superoxide generated locally by the tumor.
AML is primarily a disease of the elderly. The Swedish Adult Acute Leukemia Registry reported that the highest incidence of AML occurred between the ages 80 and 85 years, with a median age at diagnosis of 72 years.12 Outcomes for older patients with AML are poor. However, this poor prognosis is not sufficiently explained by more frequent adverse prognostic factors or genetic complexities of the tumor.12 In a comparison between younger and older patients enrolled in the UK Medical Research Council’s 10th AML trial (age <55 years at diagnosis)26 and AML trial 11 (aged 60+ years),27 Grimwade et al found that the poorer survival of older patients with favorable cytogenetic abnormalities, compared with younger individuals with similar genetic lesions, could not be accounted for by the frequency, nature, or number of secondary aberrations. Rather, the deleterious effect of advancing age at time of diagnosis remained a highly significant prognostic factor even after hierarchical cytogenetic risk group was taken into account.28 The causes for the poorer prognosis of older compared with younger patients with similar leukemia cell biologic features are likely, but many are not yet completely explained. These observations are consistent however with the hypothesis that biological differences in the older vs younger tumor host environment contribute to poorer outcomes in older patients. Our findings show that AML induces senescence in the BM microenvironment of the host and that this in turn was protumoral. We therefore hypothesize that a senescent microenvironment (which also occurs with aging) may contribute to these clinical observations.
We observed that both p16INK4a and p21 are upregulated in BMSC from mice engrafted with human AML cells. Other studies showed that mice with single p16INK4a or p21 knockout are still capable of senescence because of compensatory actions of p16INK4a and p21; however, cells from double knockout mice show little or no senescence.16,29 Using the p16-3MR mouse model to generate an in vivo model of the mouse AML microenvironment, we show that induction of p16INK4a expression in BMSC was sufficient to generate senescence and promote progression AML tumor progression. Knockdown of p21 had no significant effect on AML tumor volume in our studies.
Other in vivo models of p16INK4a senescence20,21 showed that expression of p16INK4a with age does not necessarily predict overall cancer development, suggesting that the accumulation of senescent cells is not a principal determinant of cancer-related death. In these studies, p16INK4a activation was observed in emerging neoplasms and in surrounding stromal cells in 5/5 solid tumor models studied. Such data suggest that, although it may not be sufficient to cause malignancy, p16INK4a activation is a characteristic of emerging cancer. Conversely, others have shown that the elimination of senescent cells during aging in mice produces smaller and presumably less aggressive tumors,30 and that cancer-associated fibroblasts from breast tumors have a proliferative phenotype and associated low p16INK4a expression compared with noncancer-associated fibroblasts.31 Thus, the presence of senescent stromal cells may be tumor specific. Here we add AML to the list of diseases in which p16INK4a activation in cancer associated stromal cells is protumoral.
Using p16-3MR mice, we show that AML induces a senescent BM phenotype through superoxide derived from the tumor. ROS are known to induce senescence in both benign and malignant tissue.9,11,22 Moreover, most cancers have been shown to produce ROS.32 We and others showed that NOX2-derived superoxide drives part of the malignant phenotype of AML.24,25 Because the known stimuli of cellular senescence include DNA damage and ROS,33,34 we investigated whether SASP in the BM microenvironment, occurring in response to AML, was a consequence of AML-induced superoxide. We found that ROS drives BM cellular senescence through BMSC p16INK4a expression. Furthermore, using p16-3MR mice, we showed that targeting NOX2 impaired BMSC senescence and leukemic proliferation.
Cellular senescence is emerging as a fundamental and complex component of both health and disease.35 Senescence is a response to cellular damage, which results in irreversible cell-cycle arrest and subsequent secretion of cytokines, chemokines, and other factors known as the SASP.11,36 The beneficial functions of senescence include the promotion of tissue repair, wound healing, and tumor suppression.16,37,38 In addition, senescent cells naturally accumulate with age and contribute to the pathophysiology of a spectrum of age-related disorders including cancers, atherosclerosis, and osteoarthritis.36 In an apparent paradox, it is increasingly recognized that cellular senescence underpins a number of both degenerative and hyperplastic diseases of aging.7 Therapeutically, clearance of senescent cells from the microenvironment has been reported to attenuate the effects of age-related degenerative illness and result in a regenerative program of healing in a model of osteoarthritis.39 Such data promote the hypothesis that drug targeting of senescent cells might improve current treatments (and outcomes) more broadly for patients with degenerative disorders. Similarly, the data presented here, and by others,40 linking senescent cells in the leukemia microenvironment to the malignant phenotype of AML, suggest that chemotherapeutic strategies, which include targeting of senescent cells in the microenvironment, may benefit patients with leukemia either through direct effects on tumor development and/or by enhancing responses to tumor directed chemotherapy drugs.
P16INK4a regulates the cell cycle by slowing progression from G1 to S phase. In 1 enforced expression model of p16INK4a, several components of the SASP were independent of p16INK4a expression, so several aspects of the SASP are considered a damage response that is separable from the growth arrest.41 Nevertheless, both the SASP and growth arrest occurred as a consequence of ROS-mediated p16INK4a activation.41 Here we show that p16INK4a-driven BMSC senescence is a consequence of increased ROS generated by malignant cells. Whether the SASP component of senescence is directly down stream of p16INK4a or mediated through alternate pathways in response to ROS is not clear. It is, however, the case that deletion of p16INK4a expressing senescent cells in the tumor microenvironment directly inhibits AML growth and improves animal survival.
In summary, our results describe the protumoral pathophysiologic consequences of leukemia-induced cellular senescence in the BM microenvironment and the mechanism by which this occurs. Our data open the door to future studies investigating the utility of compounds, not targeting the malignant cell, but targeting the “benign” senescent cells that surround and support the tumor.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Richard Ball, Mark Wilkinson, and Iain Sheriffs, Norwich Biorepository (United Kingdom), for help with sample collection and storage. The authors also thank Allyson Tyler, Ian Thirkettle, and Karen Ashurst from the Laboratory Medicine Department at the Norfolk and Norwich University Hospital for technical assistance.
This research was supported by the Norwich Research Park, Ministry of Higher Education and Scientific Research of the State of Libya, The Big C cancer charity (Norfolk, United Kingdom), The Rosetrees Trust, The National Institute for Health Research (United Kingdom), the US National Institutes of Health (National Institute on Aging), and the Larry L. Hillblom Foundation.
Authorship
Contribution: A.M.A.-A., C.H., Y.S., K.M.B., and S.A.R. designed the research; A.M.A.-A., C.H., Y.S., C.R.M., R.E.P., I.M., E.F., J.M., A.M., and M.S.S. performed the research; S.A.R., C.H., Y.S., C.R.M., and R.E.P. carried out in vivo work; J.C., T.M., F.D.P., C.J.I., A.C., A.H., and K.M.B. provided essential reagents and knowledge; and A.M.A.-A., C.H., Y.S., K.M.B., and S.A.R. wrote the paper, with edits from J.C.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Stuart A. Rushworth, Department of Molecular Haematology, Norwich Medical School, Norwich Research Park, Norwich, NR4 7UQ, United Kingdom; e-mail: s.rushworth@uea.ac.uk; and Kristian M. Bowles, Department of Molecular Haematology, Norwich Medical School, Norwich Research Park, Norwich, NR4 7UQ, United Kingdom; e-mail: k.bowles@uea.ac.uk.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal