

Key Points
α-Defs accelerate fibrin polymerization, increase fiber density and branching, and impede fibrinolysis in vitro.
Transgenic mice expressing human α-def-1 developed larger and more occlusive venous clots resistant to heparin and fibrinolysis.

Abstract
Inflammation and thrombosis are integrated, mutually reinforcing processes, but the interregulatory mechanisms are incompletely defined. Here, we examined the contribution of α-defensins (α-defs), antimicrobial proteins released from activated human neutrophils, on clot formation in vitro and in vivo. Activation of the intrinsic pathway of coagulation stimulates release of α-defs from neutrophils. α-Defs accelerate fibrin polymerization, increase fiber density and branching, incorporate into nascent fibrin clots, and impede fibrinolysis in vitro. Transgenic mice (Def++) expressing human α-Def-1 developed larger, occlusive, neutrophil-rich clots after partial inferior vena cava (IVC) ligation than those that formed in wild-type (WT) mice. IVC thrombi extracted from Def++ mice were composed of a fibrin meshwork that was denser and contained a higher proportion of tightly packed compressed polyhedral erythrocytes than those that developed in WT mice. Def++ mice were resistant to thromboprophylaxis with heparin. Inhibiting activation of the intrinsic pathway of coagulation, bone marrow transplantation from WT mice or provision of colchicine to Def++ mice to inhibit neutrophil degranulation decreased plasma levels of α-defs, caused a phenotypic reversion characterized by smaller thrombi comparable to those formed in WT mice, and restored responsiveness to heparin. These data identify α-defs as a potentially important and tractable link between innate immunity and thrombosis.
Introduction
Inflammation and hemostasis or thrombosis are integrated processes.1-4 Fibrin limits dissemination of microbes during implementation of the innate and adaptive immune response.5,6 Infection, inflammation and autoimmune disorders also increase the risk of thrombosis,7-11 but the pathways that link these processes are incompletely understood.
Inflammation activates the contact pathway of coagulation by releasing bacterial and platelet polyphosphates,12,13 RNA,14 DNA,15 and sulfatides16 when blood is exposed to microbial surfaces,17 collagen,18 and neutrophil extracellular traps19 and on the surfaces of intravascular devices.20 Mice lacking individual contact factors are less susceptible to developing thrombi in these settings.21-23 Diverse approaches to impairing contact activation (eg, through the use of inhibitory antibodies to factor XIIa24 and artificial antisense oligonucleotides to factor XI25,26 ) have provided thromboprophylaxis without increasing blood loss in some experimental and surgical27 settings. These data suggest that novel approaches may be required to mitigate the risk of thrombosis induced by inflammatory pathways and exposed surfaces.
Neutrophils, a principal component of the innate immune system, contribute to the increased risk, severity, and adverse outcome of thrombosis,28,29 including stroke,30-33 by contributing to the rupture of atherosclerotic plaque (reviewed by Soehnlein34 ), promoting platelet activation,35 possible carriage of tissue factor,36,37 impairing the antithrombotic function of the endothelium,36,38-40 and impeding the response to fibrinolytics,41 among other processes. A dose-dependent relationship between activated neutrophils, circulating nucleosomes, and development of deep vein thrombosis has been noted.42
Human neutrophil α-defensins (α-defs) comprise a family of four closely related peptides that constitute >5% of the total protein stored in azurophilic granules.43 The plasma concentration of α-defs rises from <15 nM in healthy individuals to as high as 50 μM after acute bacterial infection as a result of neutrophil activation.44 There is a correlation between plasma concentrations of α-defs and the incidence of myocardial infarction, stroke, and cardiovascular mortality.45-47 α-Defs bind to endothelium and vascular smooth muscle cells,48,49 deposit in atherosclerotic human coronary and carotid arteries,48,50 promote platelet activation,51,52 inhibit tissue-type plasminogen activator (tPA)–mediated fibrinolysis in vitro,53 impair lipoprotein metabolism,54 and alter vascular reactivity,55 but their involvement in thrombosis in vivo has not been established.
Investigation into the potential role of α-defs in vascular disorders such as thrombosis has been limited by the fact that unlike in humans, murine neutrophils lack these peptides.56,57 Here, we investigated the role of α-defs both in fibrin formation and stability in vitro and in the pathogenesis of thrombosis in transgenic mice expressing α-Def-1, which comprises 70% of human neutrophil α-defs. The results show that α-defs released from human neutrophils upon activation of the contact pathway of coagulation accelerate clot formation and generate compact clots with enhanced resistance to fibrinolysis in vitro and induce heparin resistance and propagate thrombus formation in vivo. Inhibition of α-def synthesis or release causes phenotypic reversion and restores responsiveness to heparin.
Methods
Materials
Recombinant tPA and dabigatran were purchased from Boehringer Ingelheim (Ingelheim am Rhein, Germany); bovine serum albumin, Tris, kaolin, and prekallikrein were from Sigma (Saint Louis, MO); tissue factor was from Perrigo (Dublin, Ireland); thrombin was from Ilex (Petah Tekva, Israel), Omrix (Jerusalem, Israel), and Sigma; rivaroxaban was from Bayer (Whippany, NJ); aprotinin was from Kamada (Peit Kama, Israel); prekallekrein-deficient plasma was from Technoclone (Vienna, Austria); and colchicine was from Rafa (Jerusalem, Israel). Active factors, XII, XI, and IX were from Kordia (Leiden, the Netherlands). Plasminogen was purified from human plasma using a tranexamic acid column from Omrix or American Diagnostica (Greenwich, CT).
Animal studies
All studies were conducted in accordance with the National Institutes of Health’s Guide for the Care and Use of Laboratory Animals and approved by the institutional animal care and use committees of the Hebrew University, Jerusalem, Israel.
Measurements of the concentrations of α-defs in human plasmas and sera
The concentrations of α-defs in human and transgenic mouse plasmas and sera were measured by enzyme-linked immunosorbent assay (ELISA) as described previously.54 See supplemental Methods (available on the Blood Web site) for experimental details.
α-Def-1 sources and radiolabeling
Two sources of α-Def-1 were employed. α-Def-1 was purchased from Peptide Institute (Osaka City, Japan). α-Def-1 was also kindly provided by Dr. Wuyan Lu (University of Maryland School of Medicine). α-Def-1 was radiolabeled with 125I as reported previously.48 See supplemental Methods for experimental details.
Binding of 125I-α-def to fibrin(ogen)
Binding of α-Def-1 to fibrinogen and fibrin was measured as described previously.48 See supplemental Methods for experimental details.
Dynamic clot turbidity
Clotting of purified fibrinogen was initiated by adding thrombin and CaCl2 in the presence or absence of α-Def-1. Fibrin formation was evaluated by monitoring the change in optical density (turbidity) at absorbance at 405 nm (A405). See supplemental Methods for experimental details.
Rate of fibrinolysis
Fibrin clots were formed as described above, but the A405 was monitored for up to 4.5 hours. Plasminogen was added to the fibrinogen before addition of thrombin and either tPA or urokinase-type plasminogen activator (uPA) in the presence or absence of α-Def-1. Because the turbidities were far higher in the presence of α-Def-1, data were normalized based on maximal turbidity. See supplemental Methods for experimental details.
Isolation of neutrophils
Immunofluorescent staining and confocal microscopy of fibrin clots
Hydrated fibrin clots formed in the presence or absence of α-Def-1 as described above were stained with an Alexa Fluor 568–conjugated anti-human fibrin mouse monoclonal antibody. Z-stacked images of immunolabeled fibrin networks were taken using a Zeiss LSM 710 laser scanning confocal microscope. Three-dimensional (3D) reconstruction, and maximal projection is shown. See supplemental Methods for experimental details.
Scanning electron microscopy
Fibrin clots formed in the absence or presence of α-Def-1 were washed, fixed, dehydrated, and sputter coated with gold-palladium as described previously.61 Triplicate samples were examined using an FEI Quanta FEG250 scanning electron microscope (FEI, Hillsboro, OR). See supplemental Methods for experimental details.
Venous thrombosis in Def++ mice
To study venous thrombosis, partial inferior vena cava (IVC) occlusion was performed62 using a model that combines external compression with a reduction in blood flow. In some experiments, the effect of IV heparin on clot formation was determined. See supplemental Methods for experimental details.
Effect of neutrophil depletion, inhibition and bone marrow transplantation (BMT) on venous thrombosis in Def++ mice
Partial IVC occlusion was performed as described previously.62 In some experiments, mice were depleted of neutrophils63 or were given colchicine in their drinking water to inhibit neutrophil degranulation54 before IVC occlusion. In other experiments, bone marrow from male Def++ mice or wild-type (WT) mice was transplanted into 6- to 8-week-old irradiated syngeneic male WT mice or bone marrow from WT mice was transplanted into Def++ mice.54 See supplemental Methods for additional details.
Statistical analysis
Group comparisons was performed using the Student t test or 1-way analysis of variance with the Newman-Keuls post hoc test.54,64 Between-group comparisons were performed using the Mann-Whitney rank test and 2-way analysis of variance with the Newman-Keuls post hoc test.64 Statistical significance was set at P < .05.
Results
Release of α-defs during blood coagulation in vitro
We first asked whether α-defs are released during clotting of whole blood in vitro. The concentrations of α-defs was over an order of magnitude higher in sera than in plasma of healthy volunteers (584 ± 85 nM vs 18 ± 3 nM, respectively; n = 10, P < .001) (Figure 1A), indicating that α-defs are released during blood coagulation in vitro.
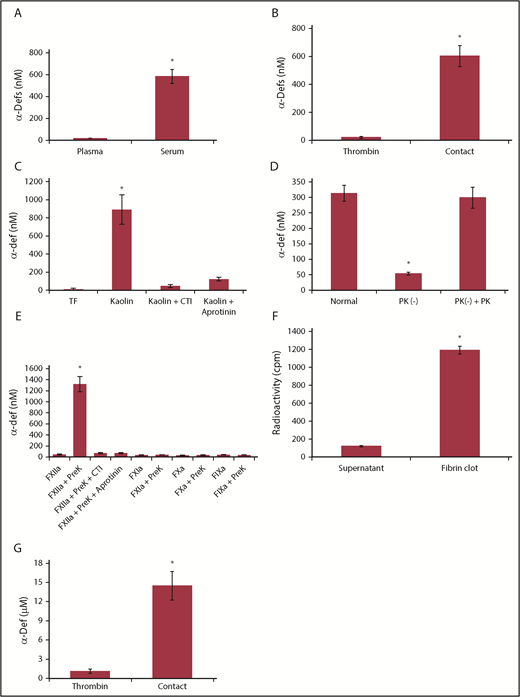
Release of endogenous α-defs during blood coagulation. (A) α-Defs in plasma and serum. Blood collected from healthy volunteers in citrate to prepare plasma was allowed to sit at room temperature for 1 hour to prepare serum. Blood was centrifuged at 1500g for 10 minutes, and the concentration of α-defs 1-3 in the plasma and serum was measured by ELISA.54 The results shown are the mean ± SD from 10 human healthy volunteers (*P < .05). (B) Role of contact activation vs thrombin. Whole blood from 11 healthy donors collected in citrate or with no anticoagulant was clotted along a glass tube or following the addition of thrombin and calcium chloride. The concentrations of α-defs measured by ELISA in sera prepared by each method were compared. The results shown are the mean ± SD in samples from 11 healthy volunteers (*P < .05). (C) Role of the intrinsic vs extrinsic pathway. Serum was generated from citrated whole blood following the addition of calcium chloride and tissue factor (TF) or kaolin, with or without aprotinin, and the concentration of α-defs was measured as in panel A. (D) Role of kallikrein. Isolated human neutrophils were added to normal plasma, prekallikrein-deficient plasma (PK (−)), or prekallikrein-deficient plasma supplemented with prekallikrein (PK (−) + PK). Clotting was initiated by adding kaolin and calcium chloride. The concentration of α-defs in the serum was measured as in panel A. The results shown are the mean ± SD of 3 experiments (*P < .05). (E) Isolated human neutrophils in phosphate-buffered saline (PBS) containing 1 mM calcium chloride were incubated with FXIIa, FXIa, FIXa, or FXa alone or together with prekallikrein (PreK) or with prekallikrein alone for 30 minutes, followed by centrifugation and separation of the supernatant fluids. In some experiments, colchicine or CTI was added along with the coagulation factors, where indicated. The concentration of α-defs in the supernatants was measured as in panel A. (F) Incorporation of α-defs into blood clots. An aliquot of 125I-α-Def-1 was added to purified fibrinogen. Clotting was induced by adding thrombin, and radioactivity in the fibrin clot and supernatant was measured. The mean ± SD of 3 experiments in shown. (G) Release of α-defs from lysed blood clots. Blood clots were formed using blood collected from healthy human volunteers as in panel B by contact with glass or by adding thrombin, separated by centrifugation, lysed by addition tPA, and recentrifuged, and the concentration of α-defs in the supernatants was measured. The mean ± SD of 3 experiments is shown.
Release of endogenous α-defs during blood coagulation. (A) α-Defs in plasma and serum. Blood collected from healthy volunteers in citrate to prepare plasma was allowed to sit at room temperature for 1 hour to prepare serum. Blood was centrifuged at 1500g for 10 minutes, and the concentration of α-defs 1-3 in the plasma and serum was measured by ELISA.54 The results shown are the mean ± SD from 10 human healthy volunteers (*P < .05). (B) Role of contact activation vs thrombin. Whole blood from 11 healthy donors collected in citrate or with no anticoagulant was clotted along a glass tube or following the addition of thrombin and calcium chloride. The concentrations of α-defs measured by ELISA in sera prepared by each method were compared. The results shown are the mean ± SD in samples from 11 healthy volunteers (*P < .05). (C) Role of the intrinsic vs extrinsic pathway. Serum was generated from citrated whole blood following the addition of calcium chloride and tissue factor (TF) or kaolin, with or without aprotinin, and the concentration of α-defs was measured as in panel A. (D) Role of kallikrein. Isolated human neutrophils were added to normal plasma, prekallikrein-deficient plasma (PK (−)), or prekallikrein-deficient plasma supplemented with prekallikrein (PK (−) + PK). Clotting was initiated by adding kaolin and calcium chloride. The concentration of α-defs in the serum was measured as in panel A. The results shown are the mean ± SD of 3 experiments (*P < .05). (E) Isolated human neutrophils in phosphate-buffered saline (PBS) containing 1 mM calcium chloride were incubated with FXIIa, FXIa, FIXa, or FXa alone or together with prekallikrein (PreK) or with prekallikrein alone for 30 minutes, followed by centrifugation and separation of the supernatant fluids. In some experiments, colchicine or CTI was added along with the coagulation factors, where indicated. The concentration of α-defs in the supernatants was measured as in panel A. (F) Incorporation of α-defs into blood clots. An aliquot of 125I-α-Def-1 was added to purified fibrinogen. Clotting was induced by adding thrombin, and radioactivity in the fibrin clot and supernatant was measured. The mean ± SD of 3 experiments in shown. (G) Release of α-defs from lysed blood clots. Blood clots were formed using blood collected from healthy human volunteers as in panel B by contact with glass or by adding thrombin, separated by centrifugation, lysed by addition tPA, and recentrifuged, and the concentration of α-defs in the supernatants was measured. The mean ± SD of 3 experiments is shown.
Release of α-defs is mediated by plasma kallikrein through activation of the contact factor pathway
Plasma kallikrein stimulates release of neutrophil elastase from azurophilic granules,65 where α-defs are stored.66 Therefore, we asked whether the contact factors, including kallikrein, are also responsible for releasing α-defs from neutrophils.67 Clotting of blood collected into a glass vacutainer was initiated in the absence of an anticoagulant to activate the contact pathway or by adding thrombin to bypass the contact pathway. Activating the contact pathway increased serum α-defs (530 ± 62 nM), whereas thrombin did not (22 ± 7 nM; Figure 1B). Identical results were obtained using each of the 4 sources of thrombin. Clotting initiated by adding kaolin increased α-defs in sera to the same extent (891.5 ± 162.3 nM). Release of α-defs by kaolin was inhibited by 400 KIU/mL aprotinin68 and 50 µg/mL corn trypsin inhibitor (CTI), suggesting a role for factor XIIa (FXIIa) as well69 (Figure 1C), and was proportional to the amount of kallikrein generated (supplemental Figure 1A-B), consistent with activation of the contact factor pathway. On the other hand, clotting initiated by 5 pM tissue factor did not affect α-def release (19 ± 13 nM) (Figure 1C; supplemental Figure 1B). The thrombin inhibitor dabigatran and the FXa inhibitor rivaroxaban delayed clotting but did not inhibit release of α-defs by kaolin (data not shown). Coagulation initiated by tissue factor (0.2 nM) to back-activate the contact factor pathway and generate additional kallikrein caused a small added increase in α-def release that was susceptible to inhibition by CTI (supplemental Figure 1B).
As an independent approach, neutrophils isolated from a healthy donor were added to autologous or prekallikrein-deficient plasma, and coagulation was initiated with kaolin. α-Defs were only released in the presence of endogenous or added prekallikrein (298 ± 34 nM) (Figure 1D). α-Defs were also released from isolated human neutrophils incubated for 30 minutes at 37°C with purified FXIIa plus prekallikrein, which was inhibited by CTI and aprotinin, but release was not induced by prekallikrein alone or FXIIa, FXIa, FVIIa, and FXa alone in the presence or absence of prekallikrein (Figure 1E). Identical results were seen in whole blood (supplemental Figure 2). In contrast to the effect of zymogen-activated serum, kallikrein did not cause the release of myeloperoxidase in parallel with α-defs.54 These data indicate that activation of the contact pathway initiates release of α-defs from neutrophils during clot formation.
Incorporation of α-defs into fibrin clots
We next asked whether α-defs released during clot formation are incorporated into the fibrin network. As a first approach, 125I-α-Def-1 (7.5 μM final concentration) was added to a solution of fibrinogen (3 mg/mL), and clotting was initiated by thrombin (0.033 U/mL). Clots were separated by centrifugation, and radioactivity in the clots and supernatants was measured. Approximately 90% of the radiolabeled α-Def-1 was associated with the clot (Figure 1F). We then measured the concentration of α-defs in clots formed in human blood activated by kaolin or thrombin and then released by adding tPA and plasminogen. The concentration of α-defs released from lysed blood clots formed by activating the contact pathway was 14.4 ± 1.9 μM compared with 1.1 ± 0.4 μM released from clots formed by adding thrombin (P < .05) (Figure 1G).
Effect of α-Def-1 on clot formation in vitro
We next asked whether α-defs modulate fibrin formation, structure, and susceptibility to lysis. α-Def-1 increased the rate of fibrin polymerization assessed by dynamic clot turbidity and altered fibrin structure assessed by increased maximal clot turbidity in a dose-dependent manner (Figure 2A-B). α-Def-1 also shortened the lag time to clot formation in a dose-dependent manner (7.0 ± 0.2, 1.5 ± 0.2, 3.1 ± 0.2, and <2.5 minutes in the presence of 0, 1, 2.5, and 5.0 µM α-Def-1, respectively). The same effect was observed when supernatants from isolated neutrophils activated by 100 ng/mL phorbol 12-myristate 13-acetate or FXIIa plus prekallikrein were added instead of purified α-Def-1 (supplemental Figure 3A-B). Release of α-defs by neutrophils was not induced by FXIIa alone, when cells were stimulated in the presence of CTI or aprotinin (supplemental Figure 3A-B), or by the presence of 3 mg/mL fibrinogen (data not shown).
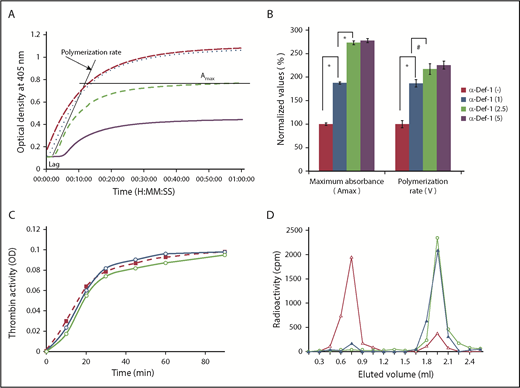
Effect of exogenous α-Def-1 on the kinetics of fibrin clot formation. (A) Effect of α-Def-1 on dynamic clot turbidity. Clotting of purified fibrinogen was initiated by adding an “activation mix” containing 0.07 U/mL human α-thrombin (see supplemental Methods for details), 0 to 10 µM α-Def-1, and 10 mM calcium chloride. Fibrin formation was evaluated by monitoring the change in turbidity (A405) in the presence of 0 µM (solid purple line), 1 µM (green dashed line), 2.5 µM (blue dotted line), and 5 µM (red dashed line) synthetic α-Def-1. One experiment representative of 3 is shown. (B) Effect of α-Def-1 on the lag time and rate of fibrin polymerization. Fibrin formation was initiated as in panel A. Lag time, rate of polymerization, and maximum absorbance (Amax) were determined in reactions containing 0 to 5 µM synthetic α-Def-1 as in panel A. The mean ± SD of 4 experiments is shown. *P < .01, #P < .05. (C) Effect of α-Def-1 on thrombin amidolytic activity. Thrombin was added to PBS containing a chromogenic substrate in the absence (red dashed line and squares) or in the presence of 2 µM (blue line and circles) or 10 µM (green line and circles) α-Def-1. One experiment representative of 3 is shown. (D) Binding of α-defs to fibrinogen and fibrin. 125I-α-Def-1 (5 µg/mL; 28 µM), twice the concentration found in fibrin clots (Figure 1G), was incubated with fibrinogen (100 µg/mL) (▲) or soluble fibrin87 (100 µg/mL) (△) in 200 μL PBS or PBS alone (○) for 60 minutes at 24°C. The mixture was loaded onto a Sephacryl S-100 gel filtration column, and radioactivity and optical density (OD) at 280 nm in each 0.5-mL fraction eluted from the column were measured. One experiment representative of 3 in shown.
Effect of exogenous α-Def-1 on the kinetics of fibrin clot formation. (A) Effect of α-Def-1 on dynamic clot turbidity. Clotting of purified fibrinogen was initiated by adding an “activation mix” containing 0.07 U/mL human α-thrombin (see supplemental Methods for details), 0 to 10 µM α-Def-1, and 10 mM calcium chloride. Fibrin formation was evaluated by monitoring the change in turbidity (A405) in the presence of 0 µM (solid purple line), 1 µM (green dashed line), 2.5 µM (blue dotted line), and 5 µM (red dashed line) synthetic α-Def-1. One experiment representative of 3 is shown. (B) Effect of α-Def-1 on the lag time and rate of fibrin polymerization. Fibrin formation was initiated as in panel A. Lag time, rate of polymerization, and maximum absorbance (Amax) were determined in reactions containing 0 to 5 µM synthetic α-Def-1 as in panel A. The mean ± SD of 4 experiments is shown. *P < .01, #P < .05. (C) Effect of α-Def-1 on thrombin amidolytic activity. Thrombin was added to PBS containing a chromogenic substrate in the absence (red dashed line and squares) or in the presence of 2 µM (blue line and circles) or 10 µM (green line and circles) α-Def-1. One experiment representative of 3 is shown. (D) Binding of α-defs to fibrinogen and fibrin. 125I-α-Def-1 (5 µg/mL; 28 µM), twice the concentration found in fibrin clots (Figure 1G), was incubated with fibrinogen (100 µg/mL) (▲) or soluble fibrin87 (100 µg/mL) (△) in 200 μL PBS or PBS alone (○) for 60 minutes at 24°C. The mixture was loaded onto a Sephacryl S-100 gel filtration column, and radioactivity and optical density (OD) at 280 nm in each 0.5-mL fraction eluted from the column were measured. One experiment representative of 3 in shown.
We next asked whether α-defs enhance clot formation by stimulating thrombin activity or whether the effects of α-defs are mediated through binding to fibrin(ogen). α-Def-1 (2 µM) did not stimulate the amidolytic activity of thrombin but rather exerted a slight inhibition at higher concentrations (10 µM) (Figure 2C). Little 125I-α-Def-1 bound directly to fibrinogen as assessed by gel filtration, whereas 92.3% of the radioactivity was found in the eluted fractions that contained soluble fibrin (Figure 2D). Together, these data suggest that α-defs accelerate fibrin formation primarily by interacting with intermediate fibrin oligomers.
Effect of α-Def-1 on clot structure in vitro
We then studied the effect of α-defs on the final structure of the fibrin network using confocal microscopy. In the presence of 5 µM α-Def-1, fibrin formed spatially nonhomogenous networks in which areas of dense and highly branched fibers were interspersed with large pores (Figure 3A). Scanning electron microscopy revealed that α-Def-1 caused bundling, entanglement, and partial conglutination of fibrin fibers resulting in the formation of a nonisotropic fibrin network with areas of accumulation and entanglement of the fibers (Figure 3B).
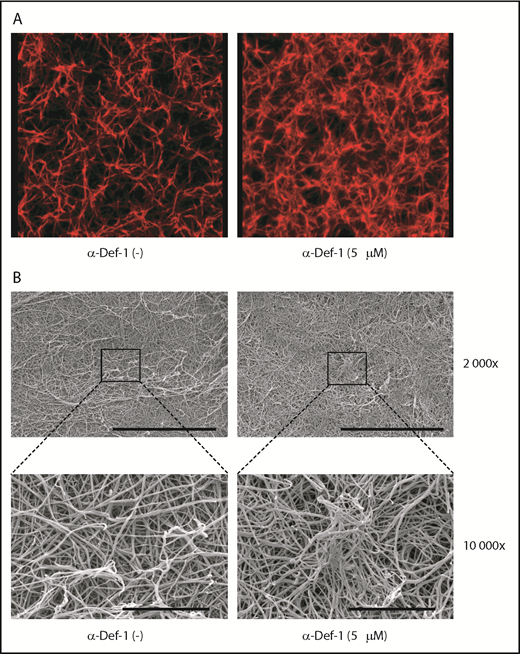
Effects of α-Def-1 on fibrin structure. (A) Representative 3D confocal microscopy images of fibrin clots formed from fibrinogen in the absence (left) or presence (right) of 5 µM α-Def-1. Fibrin was visualized using Alexa Fluor 647–labeled fibrinogen. (B) Representative scanning electron micrographs of fibrin clots formed in the absence (left) or presence (right) of 5 μM α-Def-1 at original magnification ×2000 and ×10 000. Three individual clots were studied at each experimental condition. Scale bars represent 30 μm (top) and 5 μm (bottom).
Effects of α-Def-1 on fibrin structure. (A) Representative 3D confocal microscopy images of fibrin clots formed from fibrinogen in the absence (left) or presence (right) of 5 µM α-Def-1. Fibrin was visualized using Alexa Fluor 647–labeled fibrinogen. (B) Representative scanning electron micrographs of fibrin clots formed in the absence (left) or presence (right) of 5 μM α-Def-1 at original magnification ×2000 and ×10 000. Three individual clots were studied at each experimental condition. Scale bars represent 30 μm (top) and 5 μm (bottom).
Effect of α-Def-1 on fibrinolysis in vitro
Based on these changes in fibrin structure, we assessed the impact of α-defs on tPA-induced lysis of fibrin made from purified fibrinogen in the presence of plasminogen. The time to attain 50% lysis of the fibrin clots was prolonged in the presence of increasing concentrations of α-Def-1 (Figure 4A-B). Notably, lysis was incomplete, with large pieces of fibrin remaining even after a 3-hour exposure to tPA. The amount of residual lysis-resistant fibrin was roughly proportional to the concentration of α-Def-1 (Figure 4C). The same inhibitory effect on fibrinolysis was obtained by adding supernatants from purified neutrophils activated with phorbol 12-myristate 13-acetate or FXIIa plus prekallikrein, but not by using FXIIa or prekallikrein alone, and the effect was prevented by CTI (supplemental Figure 4A). Fibrinolysis mediated by tPA, but not uPA, is inhibited when α-defs were added to preformed clots.59 In contrast, uPA-mediated lysis was blocked when α-def was added during clot formation together with plasminogen and uPA (supplemental Figure 4B). These results support the conclusion that α-defs bind to nascent fibrin (Figure 1F-G; supplemental Figure 5), altering its structure (Figure 3A-B) and susceptibility to lysis (Figure 4A-C; supplemental Figure 4A-B).
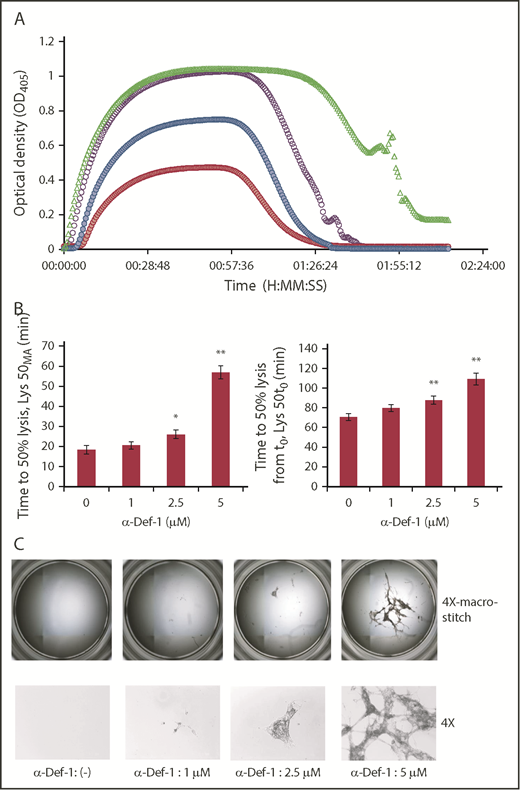
α-Def-1 delays tPA-mediated lysis of fibrin clots. (A) Fibrin was formed as in Figure 2A from fibrinogen supplemented with plasminogen, tPA, and 0-5 µM α-Def-1, and dynamic clot turbidity (A405nm) was measured. The results shown are representative of 4 experiments. (B) The time to attain 50% lysis of the fibrin clots formed in panel A, defined as the time elapsed from the maximal to the half-maximal A405 value (Lys50MA) (left) and the time from initiation of clotting needed to reduce the maximum turbidity of the clot to the half-maximal value (Lys50t0) (right), were determined. The mean ± SD is shown. The results shown are averages from 4 experiments. *P < .05, **P < .01. (C) Photographs of residual fibrin taken after lysis was allowed to proceed for 150 minutes. Images were taken with the EVOS FL Auto Cell Imaging System using EVOS software Scan and Stitch function (top). Individual representative images taken at original magnification ×4 are shown (bottom).
α-Def-1 delays tPA-mediated lysis of fibrin clots. (A) Fibrin was formed as in Figure 2A from fibrinogen supplemented with plasminogen, tPA, and 0-5 µM α-Def-1, and dynamic clot turbidity (A405nm) was measured. The results shown are representative of 4 experiments. (B) The time to attain 50% lysis of the fibrin clots formed in panel A, defined as the time elapsed from the maximal to the half-maximal A405 value (Lys50MA) (left) and the time from initiation of clotting needed to reduce the maximum turbidity of the clot to the half-maximal value (Lys50t0) (right), were determined. The mean ± SD is shown. The results shown are averages from 4 experiments. *P < .05, **P < .01. (C) Photographs of residual fibrin taken after lysis was allowed to proceed for 150 minutes. Images were taken with the EVOS FL Auto Cell Imaging System using EVOS software Scan and Stitch function (top). Individual representative images taken at original magnification ×4 are shown (bottom).
α-Def release and effect on fibrin formation and lysis ex vivo
The addition of kaolin to blood from transgenic mice (Def++) that expresses human α-Def-1 in their neutrophils56 increased the plasma concentration of α-defs from 15 ± 3 nM to 94 ± 7 nM, which was prevented by CTI and aprotinin (supplemental Figure 1). Over 90% of the α-defs released were incorporated into fibrin during clot formation (supplemental Figure 5), as seen with human blood (Figure 1G). Blood from Def++ mice clotted more rapidly, and the clots took longer to lyse (as assessed by thromboelastography) than did clots formed using blood from WT mice that do not express α-defs (Figure 5A) (R value in WT and Def++ mice, 7.4 ± 0.7 and 4.6 ± 0.6 min, respectively; n = 13 per group; P < .05). Rapid clotting in Def++ mice was preceded by an increase in α-defs in the media detected 3 minutes after the addition of kaolin (data not shown).
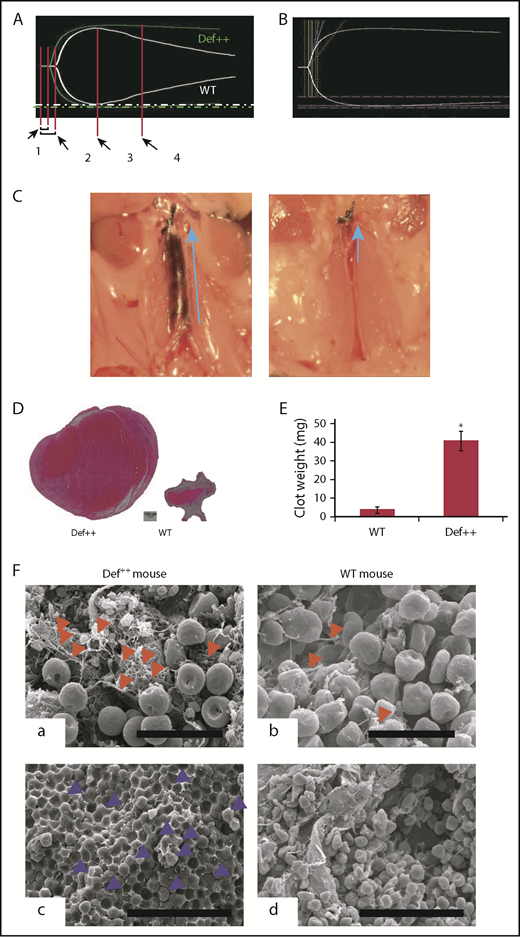
Endogenous α-defs accelerate coagulation and impair fibrinolysis in whole blood. (A) Blood was drawn from WT and Def++ mice. Clotting initiated by adding kaolin and calcium chloride was monitored by thromboelastography. “1” and “2” denote the time until the first evidence of clot formation was detected in WT and Def++ mice, respectively. “3” and “4” depict lysis in WT and Def++ mice, respectively. One experiment representative of 3 is shown. (B) Blood was drawn from a healthy human donor. Clotting was initiated and monitored as in panel A. Results show the comparability of the thromboelastography tracing in human blood and blood from Def++ mice expressing α-Def-1. One experiment representative of 3 is shown. (C) Formation of IVC thrombi in Def++ mice. Partial occlusion of the IVC was induced in Def++ and WT mice. Arrows denote the direction of blood flow and the size of the clot. (D) Three days later, clots were removed. The weights of clots extracted from Def++ and WT mice are shown. (E) The segment of the IVC containing the blood clot was excised. The clots were embedded in paraffin, sectioned, and stained with hematoxylin and eosin, and the results are shown at original magnification ×50. Panels C-E are representative of results in 12 mice. *P < .001. (F) Effect of α-def-1 on the structure of mouse IVC thrombi. Scanning electron microscopy of thrombi within the IVC of Def++ and WT mice. a and c show the clot surface, and b and d show the clot interior. Fibrin fibrils elements are identified with red arrowheads; representative compressed red blood cells (polyhedrocytes) are identified with blue arrowheads. The fibrin meshwork on the surface of the clot in Def++ is more dense (a), and many more polyhedrocytes are seen within the clot (c) than within thrombi from WT mice (b and d, respectively). Scale bars represent 10 μm (a and b) and 30 μm (c and d).
Endogenous α-defs accelerate coagulation and impair fibrinolysis in whole blood. (A) Blood was drawn from WT and Def++ mice. Clotting initiated by adding kaolin and calcium chloride was monitored by thromboelastography. “1” and “2” denote the time until the first evidence of clot formation was detected in WT and Def++ mice, respectively. “3” and “4” depict lysis in WT and Def++ mice, respectively. One experiment representative of 3 is shown. (B) Blood was drawn from a healthy human donor. Clotting was initiated and monitored as in panel A. Results show the comparability of the thromboelastography tracing in human blood and blood from Def++ mice expressing α-Def-1. One experiment representative of 3 is shown. (C) Formation of IVC thrombi in Def++ mice. Partial occlusion of the IVC was induced in Def++ and WT mice. Arrows denote the direction of blood flow and the size of the clot. (D) Three days later, clots were removed. The weights of clots extracted from Def++ and WT mice are shown. (E) The segment of the IVC containing the blood clot was excised. The clots were embedded in paraffin, sectioned, and stained with hematoxylin and eosin, and the results are shown at original magnification ×50. Panels C-E are representative of results in 12 mice. *P < .001. (F) Effect of α-def-1 on the structure of mouse IVC thrombi. Scanning electron microscopy of thrombi within the IVC of Def++ and WT mice. a and c show the clot surface, and b and d show the clot interior. Fibrin fibrils elements are identified with red arrowheads; representative compressed red blood cells (polyhedrocytes) are identified with blue arrowheads. The fibrin meshwork on the surface of the clot in Def++ is more dense (a), and many more polyhedrocytes are seen within the clot (c) than within thrombi from WT mice (b and d, respectively). Scale bars represent 10 μm (a and b) and 30 μm (c and d).
Prothrombotic effects of α-defs in vivo
We then compared outcomes in the IVC stasis model of thrombosis in Def++ and WT mice. Plasma levels of α-Def-1 rose from 17 ± 1 nM to 66 ± 5 nM after IVC occlusion in Def++ mice (n = 23; P < .05), and the clot content of α-defs was 1.44 ± 0.16 µM, which is comparable to the incorporation of α-defs in vitro (supplemental Figure 5). Every Def++ mouse developed IVC thrombi compared with 44% of WT mice (n = 13, P < .05). Clots formed in Def++ mice were overtly larger than those formed in WT mice based on their appearance in situ (Figure 5C) and visual inspection postextraction (Figure 5D-E). IVC thrombi formed in Def++ mice weighed on average ∼10-fold more than clots formed in WT mice (Figure 5D-E) (29.3 ± 3.5 vs 3.5 ± 0.6 mg; P < .001) and contained more neutrophils (591 ± 93 vs 86 ± 24 cells per field, P < .05) (Figure 5D), consistent with findings by others that α-defs promote neutrophil migration.70 Scanning electron microscopy of the fibrin meshwork of thrombi extracted from Def++ mice showed more fibrin on the clot surface (Figure 5F, a, red arrows) than was seen in thrombi removed from WT mice (Figure 5F, b), and they contained a higher proportion of tightly packed compressed polyhedral red blood cells (polyhedrocytes) within their interiors (Figure 5F, c, blue arrows), a morphologic signature of platelet-driven clot contraction,61 compared with thrombi from WT mice (Figure 5F, d).
Phenotypic conversion by preventing α-Def-1 release
We previously reported that addition of colchicine to the drinking water of Def++ mice prevented neutrophil degranulation, lowered the plasma level of α-Def-1, and prevented their pathological effect on lipoprotein metabolism.54 Therefore, we next compared thrombus development in Def++ mice given colchicine or saline in their drinking water for 2 weeks prior to inducing IVC stenosis. Pretreatment with colchicine decreased levels α-Def-1 in the circulation of Def++ mice (Figure 6A) concomitant with a decrease in the size of clots that developed in the IVC (Figure 6B), with no comparable effect on clot size in WT mice (supplemental Figure 6A-B). The fibrin network on the surface of the thrombi extracted from Def++ mice given colchicine was less dense (Figure 6C, c), and there were fewer tightly packed polyhedrocytes within the clot interior, as viewed by scanning electron microscopy (Figure 6C, d), than in thrombi from untreated animals (Figure 6C, a and b, respectively). Depletion of neutrophils in Def++ mice diminished clot size to that seen in WT mice without affecting clot size in WT mice (supplemental Figure 6A-B). Clot size was also decreased in Def++ mice given CTI prior to stenosis (supplemental Figures 7 and 8).
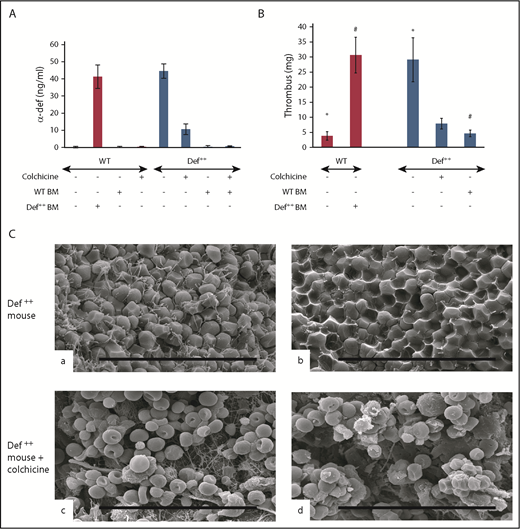
Phenotypic reversion by preventing release of α-defs. (A) Effect of colchicine and BMT on plasma α-defs. Def++ mice were given colchicine or saline in their drinking water for 2 weeks. A second cohort of WT mice underwent BMT from Def++ mice that received either colchicine or saline for 2 weeks. Plasma α-Def-1 was measured by ELISA. Results shown are the mean ± SD in 13 mice. (B) Effect of colchicine and BMT on thrombus development in Def++ mice. IVC stenosis was induced in Def++ mice given colchicine or saline in their drinking water and in Def++ and WT mice after BMT as described in panel A. Thrombus weight was measured as in Figure 4. The mean ± SD in 11 mice is shown (* and #, P < .05). (C) Effect of colchicine on IVC structure in Def++ mice. Scanning electron microscopy images of the surface (a and c) and interiors (b and d) of thrombi extracted from untreated Def++ mice (a and b) and Def++ mice given colchicine (c and d) are shown. Scale bars, 30 μm.
Phenotypic reversion by preventing release of α-defs. (A) Effect of colchicine and BMT on plasma α-defs. Def++ mice were given colchicine or saline in their drinking water for 2 weeks. A second cohort of WT mice underwent BMT from Def++ mice that received either colchicine or saline for 2 weeks. Plasma α-Def-1 was measured by ELISA. Results shown are the mean ± SD in 13 mice. (B) Effect of colchicine and BMT on thrombus development in Def++ mice. IVC stenosis was induced in Def++ mice given colchicine or saline in their drinking water and in Def++ and WT mice after BMT as described in panel A. Thrombus weight was measured as in Figure 4. The mean ± SD in 11 mice is shown (* and #, P < .05). (C) Effect of colchicine on IVC structure in Def++ mice. Scanning electron microscopy images of the surface (a and c) and interiors (b and d) of thrombi extracted from untreated Def++ mice (a and b) and Def++ mice given colchicine (c and d) are shown. Scale bars, 30 μm.
We also previously reported that transplanting bone marrow from WT mice into Def++ mice prevented formation of α-Def-1/low-density lipoprotein complexes and the development of early atherogenic changes in aortic roots in animals on a normal chow diet, whereas the converse was true when WT mice were transplanted with bone marrow from Def++ mice.54 Therefore, we used crosstransplantation as an independent method to assess the effect of α-Def-1 on IVC thrombosis. Plasma levels of α-Def-1 in WT mice transplanted with Def++ marrow reached levels seen in Def++ mice by week 4 posttransplant (15.8 ± 2.0 nM in transplanted mice vs 16.7 ± 1.3 nM in Def++ mice). Conversely, α-Def-1 became undetectable in the plasma of Def++ mice transplanted with WT BM by this time (Figure 6A). Concomitant with the rise in plasma α-Def-1, WT mice transplanted with Def++ BM developed clots similar in size to those seen in Def++ mice (Figure 6B), whereas IVC thrombi in Def++ mice transplanted with WT bone marrow were similar to those seen in WT mice (Figure 6B).
Resistance to heparin
Addition of α-defs to blood activated by kaolin attenuated the prolongation in clot time (R value) caused by heparin as measured by thromboelastography (Figure 5A). Therefore, we next asked whether endogenous release of α-Def-1 impaired the antithrombotic effectiveness of heparin in vivo. An IV bolus of saline or increasing doses of heparin or colchicine (or both) were given 1 hour after the onset of IVC stenosis and every 12 hours thereafter, and clot size was measured at 72 hours. Def++ mice required ∼20-fold more heparin than WT mice (200 U/kg vs 10 mg/kg, P < .05) to prevent clot formation (Figure 7A). IV colchicine given every 12 hours decreased clot size (Figure 7B), and coadministration with heparin reduced the dose of heparin required to prevent IVC thrombosis by ∼20-fold (200 mg/kg vs 10 mg/kg, P < .05) (Figure 7A-B) without increasing tail vein bleeding times (8.1 vs 8.2 min) (Figure 7C) or extravasation of hemoglobin following transection (0.41 vs 0.44 mg%) (Figure 7D). When Def++ mice were given a subtherapeutic dose of colchicine (0.1 mg/kg) combined with an “ineffective” dose of heparin (10 U/kg), thrombus size was reduced on average by 60% (P < .05) (Figure 7E) without an increase in tail bleeding (Figure 7E).
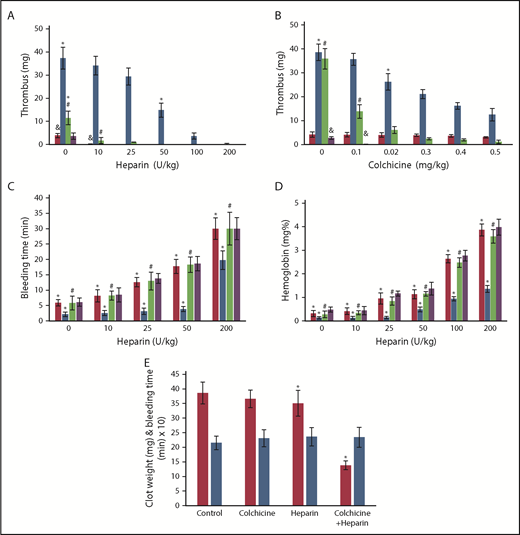
Effect of colchicine on heparin antithrombotic activity in Def++mice. (A-B) Prevention of IVC thrombosis by colchicine. (A) One hour after IVC stenosis was induced, mice were given IV saline (n = 13), heparin (0-200 U/kg) (n = 17), a fixed dose of colchicine (0.5 mg/kg) (n = 16), or heparin + colchicine (n = 21). WT mice (red) and Def++ mice (blue) were given heparin alone. WT mice (purple) and Def++ mice (green) were given heparin and colchicine. Clots were extracted and weighed 72 hours after stenosis (*, #, &, P < .05). (B) IVC stenosis was induced as in panel A. One hour later, mice were given IV saline (n = 9), colchicine (0-0.5 mg/kg) (n = 17), a fixed dose of heparin (10 U/kg) (n = 14), or heparin + colchicine (n = 14). WT mice (red) and Def++ mice (blue) were given colchicine alone. WT mice (purple) and Def++ mice (green) were given heparin and colchicine. Clots were extracted and weighed 72 hours after stenosis (*, #, P < .05). (C-D) Effect of colchicine on bleeding. (C) Mice were treated as in panel A. Tail-bleeding times were measured at 72 hours after receiving the indicated doses of heparin, colchicine, or both. WT mice (red) and Def++ mice (blue) were given heparin alone. WT mice (purple) and Def++ mice (green) were given heparin and colchicine. (D) The hemoglobin concentration in blood extravasated during the initial 30 minutes after transection. WT mice (red) and Def++ mice (blue) were given heparin alone. WT mice (purple) and Def++ mice (green) were given heparin and colchicine (*, #, P < .05). (E) Clot weight (red) and tail-bleeding times (blue) were measured 72 hours after IVC stenosis, followed 1 hour later by IV administration of colchicine (0.1 mg/kg) (n = 14), heparin (10 U/kg) (n = 14), or both (n = 11), as described in panel A. The mean ± SD are shown (*, #, P < .05).
Effect of colchicine on heparin antithrombotic activity in Def++mice. (A-B) Prevention of IVC thrombosis by colchicine. (A) One hour after IVC stenosis was induced, mice were given IV saline (n = 13), heparin (0-200 U/kg) (n = 17), a fixed dose of colchicine (0.5 mg/kg) (n = 16), or heparin + colchicine (n = 21). WT mice (red) and Def++ mice (blue) were given heparin alone. WT mice (purple) and Def++ mice (green) were given heparin and colchicine. Clots were extracted and weighed 72 hours after stenosis (*, #, &, P < .05). (B) IVC stenosis was induced as in panel A. One hour later, mice were given IV saline (n = 9), colchicine (0-0.5 mg/kg) (n = 17), a fixed dose of heparin (10 U/kg) (n = 14), or heparin + colchicine (n = 14). WT mice (red) and Def++ mice (blue) were given colchicine alone. WT mice (purple) and Def++ mice (green) were given heparin and colchicine. Clots were extracted and weighed 72 hours after stenosis (*, #, P < .05). (C-D) Effect of colchicine on bleeding. (C) Mice were treated as in panel A. Tail-bleeding times were measured at 72 hours after receiving the indicated doses of heparin, colchicine, or both. WT mice (red) and Def++ mice (blue) were given heparin alone. WT mice (purple) and Def++ mice (green) were given heparin and colchicine. (D) The hemoglobin concentration in blood extravasated during the initial 30 minutes after transection. WT mice (red) and Def++ mice (blue) were given heparin alone. WT mice (purple) and Def++ mice (green) were given heparin and colchicine (*, #, P < .05). (E) Clot weight (red) and tail-bleeding times (blue) were measured 72 hours after IVC stenosis, followed 1 hour later by IV administration of colchicine (0.1 mg/kg) (n = 14), heparin (10 U/kg) (n = 14), or both (n = 11), as described in panel A. The mean ± SD are shown (*, #, P < .05).
Discussion
Inflammation and thrombosis are closely linked processes. Fibrin contributes to the innate immune response by impeding tissue invasion by microbial pathogens,71,72 providing a provisional matrix to direct initially neutrophils and later macrophages to their targets, and stimulating cytokine release from leukocytes,73 among other impacts on the host response.74 The multiplicity of processes associated with “inflammatory thrombosis” includes pathways that are not directly responsive to antithrombotic management directed at the extrinsic pathway and generation of thrombin.2 Rapid recruitment of neutrophils to sites of thrombosis as the result of ischemic injury contributes to impaired responsiveness to fibrinolytic therapy and reperfusion injury.75 However, the mechanisms by which neutrophils enhance clot formation and impair clot resolution are incompletely defined.
The results of this study show that neutrophil-derived α-defs promote clot formation. α-Defs, at concentrations that develop under pathological conditions bind directly to nascent fibrin. Binding of α-defs accelerates fibrin polymerization, increases fibrin mass, and impairs fibrinolysis. These effects of α-defs may contribute to their antimicrobial function by accelerating the formation and enhancing the stability of the fibrin shield that acts as an early barrier against microbial infiltration.76 Moreover, the high local concentration of α-defs released rapidly following coagulation may generate high local concentrations of antimicrobial activity that precede neutrophil migration and egress into inflamed tissue.
However, α-defs incorporated into fibrin during clotting also altered clot architecture, forming a nonhomogeneous network showing areas of densification interspersed with large pores. Bundling and entanglement of fibers lead to formation of abnormal fibrin networks that have been associated previously with a prothrombotic phenotype in a number of pathological conditions, including those with a strong inflammatory component and recruitment of neutrophils.77-80 Such changes in fibrin structure influence its susceptibility to lysis and mechanical properties.81-83 Thus, the profound effect of α-defs on clot formation in vitro and in vivo can be attributed to alterations of fibrin structure described here, as well as to the previously described inhibitory effects on tPA-mediated fibrinolysis.53,59 These data help to explain how incorporation of α-defs into clots may contribute to an increased tendency for thrombus formation and incomplete fibrinolysis that might lead to incomplete vascular recanalization.84
The effects of α-defs on clot formation were also seen in vivo. α-Def-1, the most prevalent α-def in human neutrophils, is released when clotting is initiated in a transgenic mouse. α-Def-1 transgenic mice formed dramatically larger venous thrombi than WT mice. Thrombi formed in α-Def-1–expressing mice were also more resistant to dissolution by tPA. Indeed, thrombi extracted from Def++ mice contained a high proportion of tightly packed polyhedral red blood cells (polyhedrocytes),61 as revealed by scanning electron microscopy, a morphologic signature of intense clot contraction (Figure 6). The more extensive clot contraction seen in Def++ mice might retard the diffusion of lytic enzymes and thereby increase resistance to fibrinolysis.
These observations provide a newly described link between activation of the contact factor pathway during inflammation and thrombosis, and they suggest that pharmacological inhibition of thrombin (eg, by heparin) might not suffice to prevent release of α-defs and the resultant enhancement of clot stability. In our studies, clot formation was induced in the IVC in the absence of an apparent direct activation of neutrophils. Thrombus formation itself, whether through back activation of the contact factor pathway as shown here, local hypoxia, an effect of fibrin on the prothrombotic properties of the vessel wall, or vascular release of cytokines may cause sufficient release of α-defs from neutrophils to dramatically enlarge clot mass and stability. This outcome may help to explain the incomplete response to anticoagulant and fibrinolytic agents in clots invested with neutrophils1 and suggests that inhibitors of the contact factor pathway might attenuate clot formation and hasten resolution even in the absence of overt inflammation. These results also suggest that addition of drugs that inhibit the contact factor pathway or inhibit neutrophil degranulation may enhance the efficacy of inhibitors of thrombin or FXa without significantly increasing the risk of bleeding.85
Phenotypic reversion of α-def levels in plasma and thrombus formation were induced in α-Def-1 mice by BMT from WT mice and provision of colchicine in the drinking water. Colchicine has been used safely for decades to reduce neutrophil activation in patients with familial Mediterranean fever and more recently in patients with gout, osteoarthritis, or pericarditis, and a protective effect has been seen against myocardial infarction and possibly stroke.86 Def++ mice required 20-fold more heparin to prevent IVC thrombosis than WT mice. Intravenous colchicine lowered the effective anticoagulant dose of heparin to that which was effective in WT mice, with no discernable impact on hemostasis, as assessed by tail vein bleeding time or blood loss following tail transection. Additional studies will be needed to more fully understand the effect of α-defs on clot formation and structure and determine whether colchicine or other agents that prevent neutrophil degranulation provide a useful adjunctive approach to treat or prevent thrombosis that develops in the context of neutrophil activation and systemic inflammation.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Shane Lewis for measuring fibrin fiber thickness in scanning electron micrographs.
This work was supported by grants HL123912 (A.A.H. and J.W.W.), HL116916, HL142122, and HL139448 (D.B.C.), from the National Institutes of Health, National Heart, Lung, and Blood Institute; by grant 930/04 from the Israeli Science Foundation; and by the Program for Competitive Growth at Kazan Federal University.
Authorship
Contribution: A.A.-R.H. conceived the study; A.A.-R.H., R.A.-F., D.B.C., J.W.W., R.I.L., K.B., and V.S. designed experiments and analyzed data; A.A.-R.H., D.B.C., R.A.-F., J.W.W., R.I.L., and V.S. wrote the paper; R.A.-F., S.A., E.M., and M.H. performed and analyzed the experiments shown in Figures 1, 2C-D, 5, and 7A-C; and V.S., C.N., K.B., and A.R.M. performed and analyzed the experiments shown in Figures 2A-B, 3, 4, 6, and 7.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Abd Al-Roof Higazi, Department of Pathology and Laboratory Medicine, Perelman School of Medicine, University of Pennsylvania, 513A Stellar-Chance, 422 Curie Blvd, Philadelphia, PA 19104; e-mail: higazi@pennmedicine.upenn.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal