

In this issue of Blood, demonstrated that osteopontin (OPN) released by macrophages plays a key role in the chemoattraction of neutrophils in transfusion-related acute lung injury (TRALI). Their study provides a novel mechanism by which inflammation arises in this leading cause of transfusion-related death.1
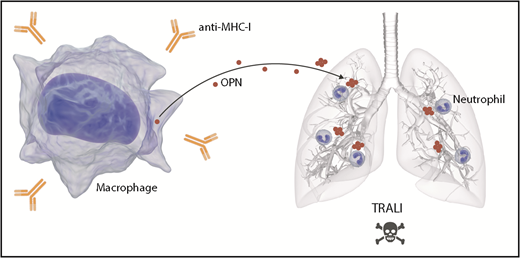
OPN mediates TRALI via stimulation of pulmonary neutrophil accumulation. The infusion of anti-MHC I antibodies triggers TRALI. OPN is released by macrophages, potentially as a result of the presence of anti-MHC I, and in its polymerized form, it accumulates in lungs. OPN in the lungs acts as a potent neutrophil chemoattractant. The efflux of neutrophils in lungs mediates TRALI.
OPN mediates TRALI via stimulation of pulmonary neutrophil accumulation. The infusion of anti-MHC I antibodies triggers TRALI. OPN is released by macrophages, potentially as a result of the presence of anti-MHC I, and in its polymerized form, it accumulates in lungs. OPN in the lungs acts as a potent neutrophil chemoattractant. The efflux of neutrophils in lungs mediates TRALI.
Although blood transfusion is a common medical procedure, it can also initiate TRALI, defined as acute respiratory distress occurring within the first 6 hours after transfusion of 1 or more units of blood or blood components. Although TRALI is rare, it is one of the main causes of death as a result of transfusion. There are no therapies available other than supportive measures such as oxygenation and ventilation.
Mechanistically, it is suggested that a combination of factors present in the blood recipient and in the blood product triggers TRALI. In this 2-hit model, there may be several reasons for the inflammation that prevails in critically ill patients: chronic alcohol abuse, smoking, liver surgery, shock, higher peak airway pressure while being mechanically ventilated, and positive fluid balance have all been incriminated as predisposing conditions in the transfused individuals (first hit).2 In the blood product, the occurrence of antibodies targeting leukocyte antigens or other soluble factors such as lysophospholipids has been suggested as the second hit.3,4 Recent studies using mouse models suggest that dendritic cells and T-regulatory cells play a protective role in the pathogenesis, but all models also incriminate neutrophils in TRALI.5 How neutrophils are recruited to the lungs has remained an enigma.
OPN is a 33-kDa glycoprotein originally identified in bone matrix and involved in the interactions between osteoblasts.6 Although its name reflects its initially identified function (osteo: bone; pontin: bridge), accumulating studies point to the involvement of OPN in inflammation,7 notably in allergic airway disease.8
The authors therefore took advantage of a mouse model of TRALI that they had previously established.5 The model requires cell priming by the injection of lipopolysaccharide and depletion of CD4+ T cells, which eliminates the protective T-regulatory cells. When antibodies targeting the major histocompatibility complex I (anti-MHC I) are infused in mice with these priming (mimicking predisposing) conditions, edema, tissue damage, and consistent accumulation of neutrophils and OPN are triggered in the lungs. Of importance, the authors demonstrated that the genetic ablation of OPN completely protected the mice from antibody-mediated TRALI.
The authors then further explored OPN in TRALI. The intravenous injection of recombinant OPN in OPN-deficient mice restored TRALI. Moreover, the blockade of OPN using anti-OPN antibodies reduced the migration of neutrophils to the lungs and protected mice from TRALI. Intriguingly, the presence of anti-OPN antibodies also led to increased levels of OPN in the blood and limited its accumulation in the lungs. These findings led the authors to hypothesize that OPN may be produced systemically and that anti-OPN antibodies may sequester the protein in blood and thereby ablate its potential chemoattractant activity.
OPN can undergo several modifications, such as phosphorylation, glycosylation, transglutamination, and thrombin-mediated fragmentation.9 Moreover, studies have confirmed that the polymerization of OPN generates a highly potent neutrophil chemoattractant.9 To verify the role of polymerization in the recruitment of neutrophils, the authors inhibited OPN polymerization in vivo. They confirmed that in the absence of polymerization, there is a reduction of neutrophil migration to the lungs and prevention of TRALI. These data suggest that OPN is released into the blood where it undergoes polymerization. Polymerized OPN then accumulates in the lungs where it is a chemoattractant for neutrophils, thereby contributing to TRALI (see figure). It is not clear how polymerization of OPN causes it to accumulate in the lungs, but it may implicate particularities of the lung vasculature. Moreover, if circulating polymerized OPN preferentially reaches the lungs and does not reach other organs, this may explain how antibody-induced organ injury in transfusion mainly affects the lungs.
Outside its expression in bone, OPN is abundantly expressed by macrophages, but not by circulating monocytes and only poorly by neutrophils. The authors therefore verified the contribution of macrophages in the TRALI model by showing that macrophage depletion blocked the migration of neutrophils to lungs and consistently protected mice from TRALI. Hence depletion strategies further determined that macrophages are the likely source of OPN in TRALI. It remains to be established how macrophages are activated in this model, but the infused anti-MHC I antibodies and the Fcγ receptors expressed by macrophages are likely contributing.10
The identification of OPN as a potential therapeutic target in TRALI is an important finding made by Kapur et al. An alternate approach might be the blockade of OPN receptor(s) on neutrophils. The authors ruled out the role of the CD44 family of receptors, which are involved in cell adhesion and migration and which indeed bind OPN. However, numerous integrins also capable of binding to OPN still remain, notably α9β1, which promotes neutrophil migration through its binding to polymerized OPN.9 The blockade of OPN polymerization or functions is unlikely to be generalizable to all transfused individuals. However, it may be applicable to transfused patients at risk of developing TRALI. Work still needs to be done to confirm that these findings are translatable to humans. Other cytokines and chemoattractants (eg, macrophage inflammatory protein-2) seem dispensable in this TRALI mouse model. Interleukin-8, a chemokine which is very potent in recruiting neutrophils in humans but absent in mice, is overexpressed in human TRALI and is a recognized risk factor.2 Thus, this work identifies OPN localization to the lungs as a novel mechanism promoting TRALI and further confirms the pathogenic role of neutrophils in this lethal pathogenesis.
Conflict-of-interest disclosure: The author declares no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal