

Key Points
At least 65% of cases of pES may be genetically determined.
Genetic findings have prognostic significance and may guide the physician’s choice of a targeted treatment.
Abstract
Evans syndrome (ES) is a rare severe autoimmune disorder characterized by the combination of autoimmune hemolytic anemia and immune thrombocytopenia. In most cases, the underlying cause is unknown. We sought to identify genetic defects in pediatric ES (pES), based on a hypothesis of strong genetic determinism. In a national, prospective cohort of 203 patients with early-onset ES (median [range] age at last follow-up: 16.3 years ([1.2-41.0 years]) initiated in 2004, 80 nonselected consecutive individuals underwent genetic testing. The clinical data were analyzed as a function of the genetic findings. Fifty-two patients (65%) received a genetic diagnosis (the M+ group): 49 carried germline mutations and 3 carried somatic variants. Thirty-two (40%) had pathogenic mutations in 1 of 9 genes known to be involved in primary immunodeficiencies (TNFRSF6, CTLA4, STAT3, PIK3CD, CBL, ADAR1, LRBA, RAG1, and KRAS), whereas 20 patients (25%) carried probable pathogenic variants in 16 genes that had not previously been reported in the context of autoimmune disease. Lastly, no genetic abnormalities were found in the remaining 28 patients (35%, the M− group). The M+ group displayed more severe disease than the M− group, with a greater frequency of additional immunopathologic manifestations and a greater median number of lines of treatment. Six patients (all from the M+ group) died during the study. In conclusion, pES was potentially genetically determined in at least 65% of cases. Systematic, wide-ranging genetic screening should be offered in pES; the genetic findings have prognostic significance and may guide the choice of a targeted treatment.
Introduction
Autoimmune diseases are numerous and common in adults.1 The underlying mechanism is complex, and involves both environmental and polygenic factors.1 A few monogenic autoimmune diseases have nevertheless been identified, mostly in children, as isolated or combined manifestations in the context of primary immunodeficiencies (PIDs).2,3 These diseases include autoimmune lymphoproliferative syndrome (ALPS, caused by mutations in the Fas apoptotic pathway),4 autoimmune polyendocrinopathy candidiasis ectodermal dystrophy (due to AIRE mutations),5 immunodysregulation polyendocrinopathy enteropathy X-linked (caused by FOXP3 mutations),6 and many others; indeed, variants in >150 different genes have been found to drive autoimmunity.7 The analysis of these genes has provided a wealth of information on the mechanisms that control reactivity to self in humans, including central negative selection, antigen receptor gene editing, regulatory T-cell count numbers and functions, peripheral elimination of autoreactive T and B cells, the clonal redemption of B cells from self-reactivity, and cis-acting regulatory elements of antigen-specific immune responses.8-11
It has been suggested that the occurrence of autoimmune disease in children (where is it relatively rare, compared with adults) may be caused by a high-risk predisposition gene. This hypothesis is based on the fact that (i) autoimmune conditions are present in relatively high proportion of PIDs (∼25%) and (ii) autoimmune conditions occur between 10 and 80 times more frequently in children with a PID than in an age-matched nonclinical population.3 For immune thrombocytopenic purpura (ITP) and autoimmune hemolytic anemia (AIHA), the relative risk factor is particularly high (up to 120 in childhood)3 ; however, no large series of nonselected patients with hematologic autoimmune disease have been comprehensively explored using next-generation sequencing (NGS). This prompted us to screen the French OBS’CEREVANCE cohort of patients with pediatric Evans syndrome (pES, a rare severe disease characterized by the simultaneous or sequential development of ITP and AIHA12,13 ) for the presence of potentially damaging mutations.
Patients and methods
Patient selection and data collection
As of 1 April 2018, a total of 203 children (ie, patients under the age of 18 years) with pES had been consecutively included in the OBS’CEREVANCE French national observational cohort. The inclusion and exclusion criteria are summarized in supplemental Table 1 (available on the Blood Web site). For each patient, relevant aspects of the family medical history, and clinical, laboratory and treatment-related data from birth to last follow-up were prospectively collected, coded, and integrated into a database. Lymphoproliferation was defined by clinically significant, persistent lymphadenopathy (>1 cm), and/or clinically significant splenomegaly detected below the costal margin in the absence of active hemolysis. Long-term follow-up data on clinical events were also collected. During the whole follow-up period, lines of treatments, treatment outcomes, and the presence of absence of additional immunopathologic manifestations and severe or recurrent infections were prospectively registered (supplemental Table 1). Written, informed consent was obtained from the patients’ parents and then (when old enough) from the patients themselves. The cohort database was registered with the French national data protection authority (CNIL) on 9 November 2009. Since 2015, 80 patients from the cohort (the study group; Figure 1) have undergone genetic testing. These 80 patients were consecutively included in the present genetic study on request by their attending physicians, once the latter had become aware of the study. Patients with a documented PID before genetic testing were excluded (n = 3). We have previously reported on a small subgroup of these patients (n = 18).14 The median (range) length of follow-up following the first observation of cytopenia and following the diagnosis of ES were 9.1 years (0.2-26.6 years) and 6.7 years (0.2-25 years), respectively. To avoid selection bias, we compared the study group of 80 patients with the 123 nontested members of the cohort (Table 1). With the exception of slightly but significantly higher frequencies of consanguinity, ITP as the first symptom, and neutropenia, there were no significant intergroup differences, notably with regard to associated immunopathologic manifestations, the median length of follow-up, the need for second-line therapy, and death.
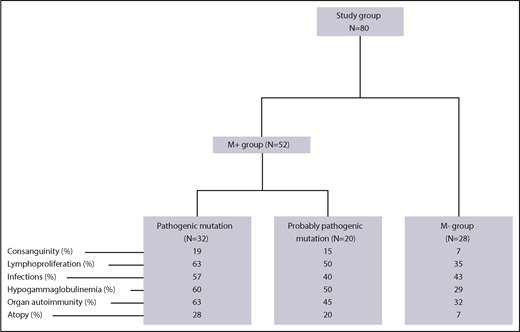
Demographical and clinical description of 203 pES patients, and the study group of 80 patients
| pES patients, N = 203 | Study group, N = 80 | Others, N = 123 | P, study group vs others | |
|---|---|---|---|---|
| Median age at first episode of cytopenia (min-max), y | 5.9 (0.2-17.4) | 5.9 (0.3-15.6) | 5.9 (0.2-17.4) | |
| Median age at ES diagnosis (min-max), y | 9.1 (0.2-19.4) | 8.7 (0.3-17.9) | 9.5 (0.2-19.4) | |
| Sex ratio (female/male) | 0.8 (91/112) | 0.68 (32/48) | 0.90 (59/64) | |
| Consanguinity, % (n) | 9 (16/177) | 13 (11) | 5 (5) | .047 |
| Immune manifestations in first-degree relatives, % (n) | 31 (57/181) | 30 (24) | 32 (33) | |
| Immune manifestations in extended relatives, % (n) | 38 (69/181) | 42 (34) | 37 (37) | |
| Occurrence of ITP and AIHA | ||||
| Simultaneous, % (n) | 42 (85) | 40 (32) | 43 (53) | |
| Sequential, % (n) | 58 (117) | 60 (48) | 57 (69) | |
| AIHA first, % (n) | 23 (47) | 14 (11) | 30 (36) | .01 |
| ITP first, % (n) | 35 (70) | 46 (37) | 27 (33) | .004 |
| Immune neutropenia, % (n) | 32 (64) | 44 (35) | 24 (29) | .002 |
| Associated immunopathologic manifestations, % (n) | 75 (153) | 82 (66) | 71 (87) | |
| Need for second-line therapy, % (n) | 72 (146) | 75 (60) | 70 (86) | |
| Rituximab, % (n) | 44 (90) | 51 (41) | 40 (49) | |
| Splenectomy, % (n) | 19 (39) | 17 (14) | 20 (25) | |
| Median follow-up from first cytopenia (min-max), y | 7.9 (0.1-29.0) | 9.1 (0.2-26.6) | 7.1 (0.1-29.0) | |
| Median follow-up from ES diagnosis (min-max), y | 5.8 (0.1-29.1) | 6.7 (0.2-25.0) | 5.5 (0.1-29.1) | |
| Deaths, % (n) | 10 (21) | 7 (6) | 12 (15) |
| pES patients, N = 203 | Study group, N = 80 | Others, N = 123 | P, study group vs others | |
|---|---|---|---|---|
| Median age at first episode of cytopenia (min-max), y | 5.9 (0.2-17.4) | 5.9 (0.3-15.6) | 5.9 (0.2-17.4) | |
| Median age at ES diagnosis (min-max), y | 9.1 (0.2-19.4) | 8.7 (0.3-17.9) | 9.5 (0.2-19.4) | |
| Sex ratio (female/male) | 0.8 (91/112) | 0.68 (32/48) | 0.90 (59/64) | |
| Consanguinity, % (n) | 9 (16/177) | 13 (11) | 5 (5) | .047 |
| Immune manifestations in first-degree relatives, % (n) | 31 (57/181) | 30 (24) | 32 (33) | |
| Immune manifestations in extended relatives, % (n) | 38 (69/181) | 42 (34) | 37 (37) | |
| Occurrence of ITP and AIHA | ||||
| Simultaneous, % (n) | 42 (85) | 40 (32) | 43 (53) | |
| Sequential, % (n) | 58 (117) | 60 (48) | 57 (69) | |
| AIHA first, % (n) | 23 (47) | 14 (11) | 30 (36) | .01 |
| ITP first, % (n) | 35 (70) | 46 (37) | 27 (33) | .004 |
| Immune neutropenia, % (n) | 32 (64) | 44 (35) | 24 (29) | .002 |
| Associated immunopathologic manifestations, % (n) | 75 (153) | 82 (66) | 71 (87) | |
| Need for second-line therapy, % (n) | 72 (146) | 75 (60) | 70 (86) | |
| Rituximab, % (n) | 44 (90) | 51 (41) | 40 (49) | |
| Splenectomy, % (n) | 19 (39) | 17 (14) | 20 (25) | |
| Median follow-up from first cytopenia (min-max), y | 7.9 (0.1-29.0) | 9.1 (0.2-26.6) | 7.1 (0.1-29.0) | |
| Median follow-up from ES diagnosis (min-max), y | 5.8 (0.1-29.1) | 6.7 (0.2-25.0) | 5.5 (0.1-29.1) | |
| Deaths, % (n) | 10 (21) | 7 (6) | 12 (15) |
max, maximum; min, minimum.
Genetic analysis
Samples of DNA were prepared from the patients’ whole peripheral blood, using standard extraction methods.15 For patients with typical clinical profiles and laboratory data suggestive of a specific PID, Sanger sequencing was performed on the most likely candidate genes. All patients with negative Sanger sequencing results (n = 69) underwent targeted NGS (tNGS) of 203 genes or (in selected cases with consanguinity or multiplex families) whole-exome sequencing. The methods used for genetic analysis and the custom panel used for tNGS are described in supplemental Appendix and supplemental Table 2, respectively. The 80 patients were then classified into 3 groups, depending on the results of the genetic analysis: (i) patients with a “pathogenic” mutation, that is, the identified mutation had been described previously in the context of autoimmune disease; (ii) patients with a “probably pathogenic” mutation, that is, the identified mutation was likely to be pathogenic but had not been described previously in the context of autoimmune disease; and (iii) patients lacking an identified genetic defect. The pathogenicity of “probably pathogenic” mutations was defined according to the following criteria: the frequency in the general population (minor allele frequency <0.01 in the GnomAD database; http://gnomad.broadinstitute.org), a literature review of the coded protein’s function, and algorithms predicting the pathogenicity of missense mutations, a Sorting Intolerant from Tolerant (SIFT) score (http://sift.jcvi.org/www/SIFT_seq_submit2.html) below 0.05 and a PolyPhen2 score (http://genetics.bwh.harvard.edu/pph2/) above 0.85, or a Combined Annotation-Dependent Depletion (CADD) score above 20 (http://cadd.gs.washington.edu/home).
Statistical analysis
Continuous variables were described as the median (range), and categorical variables were described as the number (percentage). Quantitative variables were compared using the χ2 test or (for a small sample size) the Fisher exact test. Qualitative variables were compared using the Mann-Whitney nonparametric test (for 2 variables) or an analysis of variance (for 3 variables). The percentage of patients free of additional immunopathologic manifestations was analyzed from the date of birth to the date of the first nonhematological immunopathologic manifestation or censored at the date of last follow-up. Survival estimates were calculated using the Kaplan-Meier method. Differences in survival estimates were assessed using the log-rank test. All statistical analyses were performed using R software (version 3.3.2; R Foundation for Statistical Computing, Vienna, Austria).
Results
Genetic analysis
Fifty-two of the 80 patients in the study group (65%, forming the M+ group) obtained a molecular diagnosis. Details of the mutations, the diagnostic method, and the predicted impact at the protein level are given in Tables 2 and 3. Thirty-two patients (40%) had pathogenic mutations in 9 genes known to be involved in PIDs: 23 had autosomal heterozygous germline mutations in TNFRSF6 (n = 6), CTLA4 (n = 8), STAT3 (n = 6), PIK3CD (n = 1), CBL (n = 1) or ADAR1 (n = 1), and 6 patients had autosomal homozygous disease with a homozygous LRBA mutation (n = 3), a large homozygous LRBA deletion (n = 1), or a compound heterozygous RAG1 mutation (n = 2). Three of the M+ patients had somatic heterozygous mutations, including a TNFRSF6 mutation detected in double-negative T cells (n = 1), and a KRAS mutation (n = 2). All of the variants in the “pathogenic” group have been previously described16-26 or otherwise functionally validated. TNFRSF6 variants have been validated by the observation of an apoptosis defect in T cells, with CTLA4 variants validated by low CTLA4 expression, LRBA variants validated by the absence of protein expression in a western blot analysis, and a STAT3 variant validated by elevated SOCS3 RNA transcript levels (detected using a quantitative polymerase chain reaction assay).27 Patients with RAG1 mutations developed naive T-cell lymphopenia, which was suggestive of a combined immunodeficiency.
Results of genetic testing in the study group (80 patients with pES)
| Gene (no. of patients) | Mutation type and consequences |
|---|---|
| Pathogenic mutations, n = 32, 40% | |
| TNFRSF6 (6) | Heterozygous/LOF |
| CTLA4 (8) | Heterozygous/LOF |
| STAT3 (6) | Heterozygous/GOF |
| PIK3CD (1) | Hetezozygous/GOF |
| CBL (1) | Heterozygous/LOF |
| ADAR1 (1) | Heterozygous/LOF |
| LRBA (4) | Homozygous/LOF |
| RAG 1 (2) | Compound heterozygous/LOF |
| TNFRSF6 somatic (1) | Heterozygous/LOF |
| KRAS somatic (2) | Heterozygous/GOF |
| Probably pathogenic mutations, n = 20, 25% | |
| Immune cell receptors | |
| IFNAR1 (1) | Homozygous/likely LOF |
| TNFR2 (1) | Heterozygous/likely GOF |
| TGFBR2 (1) | Heterozygous/likely LOF |
| Intracellular signaling | |
| JAK1 (2) | Heterozygous/likely GOF |
| JAK2 (1) | Heterozygous/likely GOF |
| PLCG2 (1) | Heterozygous/likely GOF |
| TRAF3 (1) | Heterozygous/likely GOF |
| CARD11 (1) | Heterozygous/likely GOF |
| ARHGEF4 (1) | Heterozygous/likely GOF |
| PTPN11 (1) | Heterozygous/likely GOF |
| PARP4 (1) | Compound heterozygous/likely LOF |
| Apoptosis regulation | |
| RIPK2 (2) | Heterozygous/likely LOF |
| APAF1 (1) | Heterozygous/likely GOF |
| Transcription factors | |
| IKZF1 (2) | Heterozygous/likely GOF |
| NFATC1 (2) | Heterozygous/likely GOF |
| IKZF2 (1) | Heterozygous/likely LOF |
| No genetic abnormalities, n = 28, 35% |
| Gene (no. of patients) | Mutation type and consequences |
|---|---|
| Pathogenic mutations, n = 32, 40% | |
| TNFRSF6 (6) | Heterozygous/LOF |
| CTLA4 (8) | Heterozygous/LOF |
| STAT3 (6) | Heterozygous/GOF |
| PIK3CD (1) | Hetezozygous/GOF |
| CBL (1) | Heterozygous/LOF |
| ADAR1 (1) | Heterozygous/LOF |
| LRBA (4) | Homozygous/LOF |
| RAG 1 (2) | Compound heterozygous/LOF |
| TNFRSF6 somatic (1) | Heterozygous/LOF |
| KRAS somatic (2) | Heterozygous/GOF |
| Probably pathogenic mutations, n = 20, 25% | |
| Immune cell receptors | |
| IFNAR1 (1) | Homozygous/likely LOF |
| TNFR2 (1) | Heterozygous/likely GOF |
| TGFBR2 (1) | Heterozygous/likely LOF |
| Intracellular signaling | |
| JAK1 (2) | Heterozygous/likely GOF |
| JAK2 (1) | Heterozygous/likely GOF |
| PLCG2 (1) | Heterozygous/likely GOF |
| TRAF3 (1) | Heterozygous/likely GOF |
| CARD11 (1) | Heterozygous/likely GOF |
| ARHGEF4 (1) | Heterozygous/likely GOF |
| PTPN11 (1) | Heterozygous/likely GOF |
| PARP4 (1) | Compound heterozygous/likely LOF |
| Apoptosis regulation | |
| RIPK2 (2) | Heterozygous/likely LOF |
| APAF1 (1) | Heterozygous/likely GOF |
| Transcription factors | |
| IKZF1 (2) | Heterozygous/likely GOF |
| NFATC1 (2) | Heterozygous/likely GOF |
| IKZF2 (1) | Heterozygous/likely LOF |
| No genetic abnormalities, n = 28, 35% |
GOF, gain of function; LOF, loss of function.
Detailed genetic results with in silico analysis in 52 pES
| Patients | Genetic | cDNA mutation | Protein level mutation | Zygosity | Protein function impact | Method | OMIM no. | PolyPhen2 | SIFT | CADD | MAF | GnomeAD |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Pathogenic | ||||||||||||
| P1 | TNFRSF6 germline | 335-12C>G | NA | Heterozygous | LOF | Sanger | 134637 | NA | NA | NA | 0 | 0 |
| P2 | TNFRSF6 germline | 808_815del | D269fsX277 | Heterozygous | LOF | Sanger | 134637 | NA | NA | NA | 0 | 0 |
| P3 | TNFRSF6 germline | 748C>T | R250X | Heterozygous | LOF | Sanger | 134637 | NA | NA | 41 | 0 | 0 |
| P4 | TNFRSF6 germline | 709G>C | A237P | Heterozygous | LOF | Sanger | 134637 | 1 | 0.01 | 23.6 | 0 | 0 |
| P5 | TNFRSF6 germline | 806_807del | D269fsX279 | Heterozygous | LOF | Sanger | 134637 | NA | NA | NA | 0 | 0 |
| P6 | TNFRSF6 germline | 506-2A>G | NA | Heterozygous | LOF | Sanger | 134637 | NA | NA | 22.4 | 0 | 0 |
| P7 | CTLA4 | 410C>T | P137L | Heterozygous | LOF | TNGS | 123890 | 1 | 0 | 27 | 0 | 0 |
| P8 | CTLA4 | 151C>T | R51X | Heterozygous | LOF | Sanger | 123890 | 0.999 | 0 | 36 | 0 | 0 |
| P9 | CTLA4 | 208C>T | R70W | Heterozygous | LOF | TNGS | 123890 | 0.999 | 0 | 32 | 0 | 0 |
| P10 | CTLA4 | 110-2A>G | NA | Heterozygous | LOF | TNGS | 123890 | NA | NA | 21 | 0 | 0 |
| P11 | CTLA4 | 151C>T | R51X | Heterozygous | LOF | TNGS | 123890 | 0.999 | 0 | 36 | 0 | 0 |
| P12 | CTLA4 | 316A>C | T106P | Heterozygous | LOF | Sanger | 123890 | 0.653 | 0.17 | 14.4 | 0 | 0 |
| P13 | CTLA4 | c.109 + 1092_568-512del | p.M38Kfs*22 | Heterozygous | LOF | Sanger | 123890 | NA | NA | NA | 0 | 0 |
| P14 | CTLA4 | 172T>G | C58G | Heterozygous | LOF | TNGS | 123890 | 0.998 | 0 | 23.9 | 0 | 0 |
| P15 | STAT3 | 1988 C>T | T663I | Heterozygous | GOF | TNGS | 102582 | 0.997 | 0.02 | 21.8 | 0 | 0 |
| P16 | STAT3 | 2147C>T | T716M | Heterozygous | GOF | TNGS | 102582 | 0.995 | 0.13 | 18.4 | 0 | 0 |
| P17 | STAT3 | 1261G>A | G421R | Heterozygous | GOF | TNGS | 102582 | 1 | 0 | 35 | 0 | 0 |
| P18 | STAT3 | 1255G>C | G419R | Heterozygous | GOF | TNGS | 102582 | 0.986 | 0.42 | 25.3 | 0 | 0 |
| P19 | STAT3 | 1919A>T | Y640F | Heterozygous | GOF | TNGS | 102582 | 0.999 | 0.34 | 24.2 | 0 | 0 |
| P20 | STAT3 | 2144C>T | P715L | Heterozygous | GOF | TNGS | 102582 | 1 | 0.11 | 25.6 | 0 | 0 |
| P21 | PIK3CD | 3061G>A | E1021K | Heterozygous | GOF | TNGS | 602839 | 0.999 | 0.01 | 31 | 0 | 0 |
| P22 | CBL | 1420G>A | A474T | Heterozygous | LOF | TNGS | 165360 | 0.536 | 0.06 | 25 | 0 | 0 |
| P23 | ADAR1 | 3019G>A | G1007R | Heterozygous | LOF | TNGS | 146920 | 1 | 0.05 | 34 | 0 | 0 |
| P24 | LRBA | 2450+1C>T | E789fsX792 | Homozygous | LOF | TNGS | 606453 | NA | NA | 25.9 | 0 | 0 |
| P25 | LRBA | del exon 18 to 30 | del exon 18 to 30 | Homozygous | LOF | WB | 606453 | NA | NA | NA | 0 | 0 |
| P26 | LRBA | 1691delT | L564RfsX25 | Homozygous | LOF | TNGS | 606453 | NA | NA | NA | 0 | 0 |
| P27 | LRBA | 7620_7621 insT | A2541YfsX2542 | Homozygous | LOF | TNGS | 606453 | NA | NA | NA | 0 | 0 |
| P28 | RAG1 | 110C>A | S37Y | Compound | LOF | TNGS | 179615 | 0.974 | 0 | 26.4 | 4.066 × 10−6 | 2 |
| 2657T>C | M886T | Heterozygous | LOF | 0.036 | 0.02 | 17.8 | 8.144 × 10−6 | 1 | ||||
| P29 | RAG1 | 335G>T | R112L | Compound | LOF | TNGS | 179615 | 0.953 | NA | 29.2 | 0 | 0 |
| 2259T>A | H753Q | Heterozygous | LOF | 0.978 | NA | 17.49 | 0 | 0 | ||||
| P30 | TNFRSF6 somatic | 749G>A | R250Q | Heterozygous | LOF | Sanger | 134637 | 1 | 0 | 33 | 0 | 0 |
| P31 | KRAS somatic | 37 G>T | G13C | Heterozygous | GOF | TNGS | 190070 | 1 | 0 | 33 | 0 | 0 |
| P32 | KRAS somatic | 37 G>T | G13C | Heterozygous | GOF | Sanger | 190070 | 1 | 0 | 33 | 0 | 0 |
| Probably pathogenic | ||||||||||||
| P33 | TNFR2 | c.482G>A | C161Y | Heterozygous | Likely GOF | TNGS | 191191 | 0.998 | 0 | 23.4 | 0 | 0 |
| P34 | IFNAR1* | 71C>T | A24V | Homozygous | Likely LOF | WES | 107450 | 0.286 | 0.12 | 23.2 | 8.408 × 10−6 | 2 |
| P35 | TGFBR2 | 851A>G | Y284C | Heterozygous | Likely LOF | TNGS | 190182 | 0.989 | 0 | 26.6 | 1.638 × 10−5 | 4 |
| P36 | JAK1 | c.1981G>A | p.V661M | Heterozygous | Likely GOF | TNGS | 147795 | 0.661 | 0.1 | 23.8 | 7.226 × 10−6 | 2 |
| P37 | JAK1 | c.17T>C | pI6T | Heterozygous | Likely GOF | TNGS | 147795 | 0.011 | 0.04 | 23.6 | 0 | 0 |
| P38 | JAK2 | c.980G>T | p.S327I | Heterozygous | Likely GOF | TNGS | 147796 | 0.851 | 0.06 | 24 | 1.228 × 10−5 | 3 |
| P39 | PLCG2 | 2625C>G | I875M | Heterozygous | Likely GOF | TNGS | 600220 | 0.316 | 0.06 | 22.1 | 0 | 0 |
| P40 | TRAF3 | 1591G>T | A531S | Heterozygous | Likely GOF | TNGS | 601896 | 1 | 0 | 28.3 | 0 | 0 |
| P41 | CARD11 | 3410A>T | D1137V | Heterozygous | Likely GOF | TNGS | 607210 | 0.375 | 0.04 | 24 | 1.805 × 10−5 | 5 |
| P42 | ARHGEF4 | 1328C>T | 443L | Heterozygous | Likely GOF | TNGS | 605216 | 0.995 | 0.07 | 27.5 | 0 | 0 |
| P43 | PTPN11 | 659G>A | R220H | Heterozygous | Likely GOF | TNGS | 176876 | 0.001 | 0.03 | 26.1 | 4.063 × 10−6 | 1 |
| P44 | PARP4 | 3284_3285delAG −2ex 26 | NA | Compound | Likely LOF | TNGS | 607519 | NA | NA | NA | 0 | 0 |
| 3426C>G | H1142Q | Heterozygous | Likely LOF | 0.971 | 0.04 | 17.1 | 0 | 0 | ||||
| P45 | RIPK2 | 593A>G | Y198C | Heterozygous | Likely LOF | TNGS | 603455 | 1 | 0 | 28.2 | 0 | 0 |
| P46 | RIPK2 | 698C>G | T233S | Heterozygous | Likely LOF | TNGS | 603455 | 0.751 | 0.14 | 23.3 | 0 | 0 |
| P47 | APAF1 | 677G>A | R226H | Heterozygous | Likely GOF | TNGS | 602233 | 0.999 | 0 | 34 | 8.122 × 10−6 | 2 |
| P48 | NFATC1 | c.613G>A | p.E205K | Heterozygous | Likely GOF | TNGS | 600489 | 0.629 | 0.5 | 21 | 0 | 0 |
| P49 | NFATC1 | c.1226+8G>A | NA | Heterozygous | Likely GOF | TNGS | 600489 | NA | NA | 6.6 | 8.527 × 10−6 | 1 |
| P50 | IKZF1 | 548G>A | R183H | Heterozygous | Likely GOF | TNGS | 603023 | 0.998 | 0 | 33 | 0 | 0 |
| P51 | IKZF1 | 1499A>G | Q500R | Heterozygous | Likely GOF | TNGS | 603023 | 0.154 | 0.12 | 22 | 0 | 0 |
| P52 | IKZF2 | 659A>G | N220S | Heterozygous | Likely LOF | TNGS | 606234 | 0.986 | 0.51 | 21.4 | 4.478 × 10−5 | 11 (0/110 000 in European) |
| Patients | Genetic | cDNA mutation | Protein level mutation | Zygosity | Protein function impact | Method | OMIM no. | PolyPhen2 | SIFT | CADD | MAF | GnomeAD |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Pathogenic | ||||||||||||
| P1 | TNFRSF6 germline | 335-12C>G | NA | Heterozygous | LOF | Sanger | 134637 | NA | NA | NA | 0 | 0 |
| P2 | TNFRSF6 germline | 808_815del | D269fsX277 | Heterozygous | LOF | Sanger | 134637 | NA | NA | NA | 0 | 0 |
| P3 | TNFRSF6 germline | 748C>T | R250X | Heterozygous | LOF | Sanger | 134637 | NA | NA | 41 | 0 | 0 |
| P4 | TNFRSF6 germline | 709G>C | A237P | Heterozygous | LOF | Sanger | 134637 | 1 | 0.01 | 23.6 | 0 | 0 |
| P5 | TNFRSF6 germline | 806_807del | D269fsX279 | Heterozygous | LOF | Sanger | 134637 | NA | NA | NA | 0 | 0 |
| P6 | TNFRSF6 germline | 506-2A>G | NA | Heterozygous | LOF | Sanger | 134637 | NA | NA | 22.4 | 0 | 0 |
| P7 | CTLA4 | 410C>T | P137L | Heterozygous | LOF | TNGS | 123890 | 1 | 0 | 27 | 0 | 0 |
| P8 | CTLA4 | 151C>T | R51X | Heterozygous | LOF | Sanger | 123890 | 0.999 | 0 | 36 | 0 | 0 |
| P9 | CTLA4 | 208C>T | R70W | Heterozygous | LOF | TNGS | 123890 | 0.999 | 0 | 32 | 0 | 0 |
| P10 | CTLA4 | 110-2A>G | NA | Heterozygous | LOF | TNGS | 123890 | NA | NA | 21 | 0 | 0 |
| P11 | CTLA4 | 151C>T | R51X | Heterozygous | LOF | TNGS | 123890 | 0.999 | 0 | 36 | 0 | 0 |
| P12 | CTLA4 | 316A>C | T106P | Heterozygous | LOF | Sanger | 123890 | 0.653 | 0.17 | 14.4 | 0 | 0 |
| P13 | CTLA4 | c.109 + 1092_568-512del | p.M38Kfs*22 | Heterozygous | LOF | Sanger | 123890 | NA | NA | NA | 0 | 0 |
| P14 | CTLA4 | 172T>G | C58G | Heterozygous | LOF | TNGS | 123890 | 0.998 | 0 | 23.9 | 0 | 0 |
| P15 | STAT3 | 1988 C>T | T663I | Heterozygous | GOF | TNGS | 102582 | 0.997 | 0.02 | 21.8 | 0 | 0 |
| P16 | STAT3 | 2147C>T | T716M | Heterozygous | GOF | TNGS | 102582 | 0.995 | 0.13 | 18.4 | 0 | 0 |
| P17 | STAT3 | 1261G>A | G421R | Heterozygous | GOF | TNGS | 102582 | 1 | 0 | 35 | 0 | 0 |
| P18 | STAT3 | 1255G>C | G419R | Heterozygous | GOF | TNGS | 102582 | 0.986 | 0.42 | 25.3 | 0 | 0 |
| P19 | STAT3 | 1919A>T | Y640F | Heterozygous | GOF | TNGS | 102582 | 0.999 | 0.34 | 24.2 | 0 | 0 |
| P20 | STAT3 | 2144C>T | P715L | Heterozygous | GOF | TNGS | 102582 | 1 | 0.11 | 25.6 | 0 | 0 |
| P21 | PIK3CD | 3061G>A | E1021K | Heterozygous | GOF | TNGS | 602839 | 0.999 | 0.01 | 31 | 0 | 0 |
| P22 | CBL | 1420G>A | A474T | Heterozygous | LOF | TNGS | 165360 | 0.536 | 0.06 | 25 | 0 | 0 |
| P23 | ADAR1 | 3019G>A | G1007R | Heterozygous | LOF | TNGS | 146920 | 1 | 0.05 | 34 | 0 | 0 |
| P24 | LRBA | 2450+1C>T | E789fsX792 | Homozygous | LOF | TNGS | 606453 | NA | NA | 25.9 | 0 | 0 |
| P25 | LRBA | del exon 18 to 30 | del exon 18 to 30 | Homozygous | LOF | WB | 606453 | NA | NA | NA | 0 | 0 |
| P26 | LRBA | 1691delT | L564RfsX25 | Homozygous | LOF | TNGS | 606453 | NA | NA | NA | 0 | 0 |
| P27 | LRBA | 7620_7621 insT | A2541YfsX2542 | Homozygous | LOF | TNGS | 606453 | NA | NA | NA | 0 | 0 |
| P28 | RAG1 | 110C>A | S37Y | Compound | LOF | TNGS | 179615 | 0.974 | 0 | 26.4 | 4.066 × 10−6 | 2 |
| 2657T>C | M886T | Heterozygous | LOF | 0.036 | 0.02 | 17.8 | 8.144 × 10−6 | 1 | ||||
| P29 | RAG1 | 335G>T | R112L | Compound | LOF | TNGS | 179615 | 0.953 | NA | 29.2 | 0 | 0 |
| 2259T>A | H753Q | Heterozygous | LOF | 0.978 | NA | 17.49 | 0 | 0 | ||||
| P30 | TNFRSF6 somatic | 749G>A | R250Q | Heterozygous | LOF | Sanger | 134637 | 1 | 0 | 33 | 0 | 0 |
| P31 | KRAS somatic | 37 G>T | G13C | Heterozygous | GOF | TNGS | 190070 | 1 | 0 | 33 | 0 | 0 |
| P32 | KRAS somatic | 37 G>T | G13C | Heterozygous | GOF | Sanger | 190070 | 1 | 0 | 33 | 0 | 0 |
| Probably pathogenic | ||||||||||||
| P33 | TNFR2 | c.482G>A | C161Y | Heterozygous | Likely GOF | TNGS | 191191 | 0.998 | 0 | 23.4 | 0 | 0 |
| P34 | IFNAR1* | 71C>T | A24V | Homozygous | Likely LOF | WES | 107450 | 0.286 | 0.12 | 23.2 | 8.408 × 10−6 | 2 |
| P35 | TGFBR2 | 851A>G | Y284C | Heterozygous | Likely LOF | TNGS | 190182 | 0.989 | 0 | 26.6 | 1.638 × 10−5 | 4 |
| P36 | JAK1 | c.1981G>A | p.V661M | Heterozygous | Likely GOF | TNGS | 147795 | 0.661 | 0.1 | 23.8 | 7.226 × 10−6 | 2 |
| P37 | JAK1 | c.17T>C | pI6T | Heterozygous | Likely GOF | TNGS | 147795 | 0.011 | 0.04 | 23.6 | 0 | 0 |
| P38 | JAK2 | c.980G>T | p.S327I | Heterozygous | Likely GOF | TNGS | 147796 | 0.851 | 0.06 | 24 | 1.228 × 10−5 | 3 |
| P39 | PLCG2 | 2625C>G | I875M | Heterozygous | Likely GOF | TNGS | 600220 | 0.316 | 0.06 | 22.1 | 0 | 0 |
| P40 | TRAF3 | 1591G>T | A531S | Heterozygous | Likely GOF | TNGS | 601896 | 1 | 0 | 28.3 | 0 | 0 |
| P41 | CARD11 | 3410A>T | D1137V | Heterozygous | Likely GOF | TNGS | 607210 | 0.375 | 0.04 | 24 | 1.805 × 10−5 | 5 |
| P42 | ARHGEF4 | 1328C>T | 443L | Heterozygous | Likely GOF | TNGS | 605216 | 0.995 | 0.07 | 27.5 | 0 | 0 |
| P43 | PTPN11 | 659G>A | R220H | Heterozygous | Likely GOF | TNGS | 176876 | 0.001 | 0.03 | 26.1 | 4.063 × 10−6 | 1 |
| P44 | PARP4 | 3284_3285delAG −2ex 26 | NA | Compound | Likely LOF | TNGS | 607519 | NA | NA | NA | 0 | 0 |
| 3426C>G | H1142Q | Heterozygous | Likely LOF | 0.971 | 0.04 | 17.1 | 0 | 0 | ||||
| P45 | RIPK2 | 593A>G | Y198C | Heterozygous | Likely LOF | TNGS | 603455 | 1 | 0 | 28.2 | 0 | 0 |
| P46 | RIPK2 | 698C>G | T233S | Heterozygous | Likely LOF | TNGS | 603455 | 0.751 | 0.14 | 23.3 | 0 | 0 |
| P47 | APAF1 | 677G>A | R226H | Heterozygous | Likely GOF | TNGS | 602233 | 0.999 | 0 | 34 | 8.122 × 10−6 | 2 |
| P48 | NFATC1 | c.613G>A | p.E205K | Heterozygous | Likely GOF | TNGS | 600489 | 0.629 | 0.5 | 21 | 0 | 0 |
| P49 | NFATC1 | c.1226+8G>A | NA | Heterozygous | Likely GOF | TNGS | 600489 | NA | NA | 6.6 | 8.527 × 10−6 | 1 |
| P50 | IKZF1 | 548G>A | R183H | Heterozygous | Likely GOF | TNGS | 603023 | 0.998 | 0 | 33 | 0 | 0 |
| P51 | IKZF1 | 1499A>G | Q500R | Heterozygous | Likely GOF | TNGS | 603023 | 0.154 | 0.12 | 22 | 0 | 0 |
| P52 | IKZF2 | 659A>G | N220S | Heterozygous | Likely LOF | TNGS | 606234 | 0.986 | 0.51 | 21.4 | 4.478 × 10−5 | 11 (0/110 000 in European) |
CADD, Combined Annotation-Dependent Depletion; cDNA, complementary DNA; MAF, minor allele frequency; NA, not applicable; OMIM, Online Mendelian Inheritance in Man; SIFT, Sorting Intolerant from Tolerant; TNGS, targeted next-generation sequencing; WB, western blot; WES, whole-exome sequencing. See Table 2 for expansion of other abbreviations.
Patient PBMCs were shown to have impaired induction of interferon-stimulated genes (n = 6) after stimulation by IFNα comparatively to control cells.
Twenty patients (25%) had “probably pathogenic” variants in 16 genes that had not previously been described in the context of autoimmune diseases and/or were suggestive of new molecular mechanisms (Tables 2 and3). All but one of the variants met our predicted pathogenicity criteria (a CADD score >20, or PolyPhen2 and SIFT scores >0.85 and <0.05, respectively). The NFATC1 variant P49 had a lower CADD score but had a demonstrated impact on exon splicing with complementary DNA (cDNA; data not shown). Furthermore, another patient (P48) had a pathogenic variant in NFATC1. No other variants in immune-related genes were detected in these 20 patients. These variants mainly involve immune cell receptors (with a probable loss of function [LOF] in TNFR2, TRAF3, IFNAR1, and TGFBR2, n = 1 for each), intracellular signaling (JAK1, with a probable gain of function [GOF] in 2 cases, and JAK2 with a probable GOF, PLCG2 with a probable GOF, CARD11 with a probable GOF, PARP4 with a probable LOF, ARGHEF4 with a probable GOF, and PTPN11 with a probable GOF, n = 1 for each), the regulation of apoptosis (RIPK2 with a probable LOF in 2 cases, and APAF1, with a probable LOF in 1 case), and lastly, transcriptional factors in immune cells (IKZF1 with a probable GOF and NFATC1 with a probable GOF in 2 cases each, and IKZF2 with a probable LOF in 1 case).
In light of recently published results, some of the variants merit further comment. IKZF1 LOF mutations have been described in patients with common variable immunodeficiency (haploinsufficiency mutations)28 and combined immunodeficiency (dominant-negative mutations).29 In contrast, we suggest that the 2 patients studied here carry a previously undescribed GOF mutation leading to an autoimmune phenotype; this GOF hypothesis is supported by preliminary data, that is, greater binding of Ikaros to specific DNA targets in an electrophoretic mobility shift assay.30 Various types of CARD11 mutation can lead to distinct phenotypes; for example, biallelic null mutations lead to severe T- and B-cell immune deficiencies,31 whereas germline GOF mutations give rise to “B-cell expansion with NF-κB and T-cell anergy” (BENTA) disease.32 Lastly, hypomorphic/dominant-negative mutations predispose to atopic phenotypes and variable immunodeficiency.33,34 The present CARD11 mutation has never been reported; functional data are not yet available, and the phenotype differs from those reported. A recent study indicated that hypomorphic CARD11 mutations are associated with a variety of immunologic phenotypes and (in some cases) atopic disease,35 including autoimmunity in 20% of patients. PLCG2 deletions are responsible for the PLCG2-associated antibody deficiency and immune dysregulation (PLAID) syndrome (cold urticaria, immunodeficiency, and autoimmunity36 ), and missense GOF mutations have been associated with a dominantly inherited autoinflammatory disease with immunodeficiency.37,38 The PLCG2 mutation (I875M) reported here has never been described. The fact that it is located near to the mutation reported by Neves et al38 (L848P) prompts us to suspect a GOF mutation. JAK1 GOF mutations have been recently described in patients with severe atopic dermatitis and hypereosinophilic syndrome without autoimmune manifestations.39 Given the importance of the JAK-STAT pathway in PIDs with autoimmunity (such as STAT1 and STAT3 GOF mutations), a GOF mutation leading to hyperactivation of the JAK-STAT pathway and a specific clinical phenotype is likely. Germline JAK2 mutations have previously been linked to myeloproliferative neoplasms but not to autoimmune manifestations. By analogy with the JAK1 variants, we suggest that a JAK2 GOF mutation is present. Homozygous IFNAR1 mutations have been recently described in patients with increased susceptibility to viral infections40 ; our patient’s clinical phenotype (ie, autoimmunity) is quite different. Preliminary data have evidenced the impaired induction of interferon-stimulated genes (n = 6) in the patient’s peripheral blood mononuclear cells (PBMCs; relative to PBMCs from healthy controls) following stimulation with interferon-α, thus suggesting a LOF mutation (data not shown). Germline mutations in PTPN11 are usually associated with Noonan syndrome, which is characterized by a dysmorphic syndrome, congenital heart disease, and coagulation defects.41 However, dysregulation of the MAPK pathway due to a somatic mutation in NRAS has been linked to an ALPS-like syndrome (RAS-associated ALPS-like disease [RALD25,42 ]). We suggest that a GOF mutation in PTPN11 in the present case might lead to hyperactivation of the MAPK pathway and thus the onset of autoimmunity. TGFBR2 mutations have been linked to Marfan syndrome–related disorders.43 The patient described here does not present a Marfan phenotype. A LOF mutation that impairs regulatory T-cell activity could conceivably lead to autoimmunity. All of the other germline mutations detected in the “probably pathogenic” group have never been described in human diseases; all are now undergoing extensive functional validation, in order to precisely characterize the mechanism leading to autoimmunity.
Lastly, neither tNGS (n = 20) nor whole-exome sequencing (n = 8) detected any genetic abnormalities in 28 of the 80 patients (35%, forming the M− group).
Clinical phenotypes and outcomes
The clinical presentations of the 80 children in the study group are described in detail in Table 1. The median (range) duration of follow-up was 9.1 years (0.2-26.6 years). pES was associated with additional immunopathologic manifestations in 66 of the patients (82%). These manifestations were present before the first episode of cytopenia in 18 cases (27%, with a median [range] time interval of 3.6 years [0.4-14 years]), and arose at the same time as the first episode of cytopenia in 22 cases (33%), or arose afterward in 26 cases (39%, with a median [range] time interval of 3 years [0.1-11.5]). Forty of the 80 patients (50%) had lymphoproliferation (8 had isolated splenomegaly, 8 had isolated lymph node enlargement, and 24 had both), 37 (46%) had hypogammaglobulinemia (after anti-CD20 antibody treatment in 25 of these), and 38 (47%) had various autoimmune/autoinflammatory manifestations (mainly liver, digestive tract, and lung manifestations). The median (range) number of additional immunopathologic manifestations per patient was 2 (0-8). Severe or recurrent infections occurred in 47% of the patients, although we were not able to determine whether these were disease-related or treatment-related.
We next compared the M+ and M− groups with regard to the phenotype (Figure 1; Table 4). The M+ group was more likely to present with additional immunopathologic manifestations (P = .007) (Figure 2) and hypogammaglobulinemia (P = .02). There was a nonsignificant trend toward a greater frequency of lymphoproliferation (P = .06) in the M+ group. No significant differences were observed between the patients with a known pathogenic mutation and the patients with a probable pathogenic variant, with the exception of the occurrence of gut immunopathologic manifestations (P = .008) and a trend toward the more frequent occurrence of lung manifestations (P = .07) and ALPS biomarkers (P = .07) in the “pathogenic” group (Table 4). Overall, these results suggest that pES tends to be more severe when it occurs in the context of genetic abnormalities, as illustrated by a higher median number of lines of treatment. The time to the first immunopathologic manifestation was shorter in the M+ group than in the M− group (Figure 3), while the shape of the curves suggests that the genetic impact on the occurrence of additional immunopathologic manifestations is greater in the first 5 years of life than later in life. Six of the 80 patients in the study group died during the study period (supplemental Figure 1; supplemental Table 3). It is noteworthy that all 6 belonged to the M+ group. Death occurred at a median (range) age of 18.9 years (3.9-25 years), and the median (range) time interval between the initial diagnosis and death was 10.7 years (0.9-24.6 years). Three deaths were disease-related (cerebral hemorrhage, juvenile myelomonocytic leukemia, and fulminant hepatitis) and 3 were disease- or treatment-related (sepsis, Epstein-Barr virus lymphoproliferation, and pneumococcal meningitis). Three asplenic patients died, although asplenic sepsis was the cause of death in only 1 patient.
Clinical presentation of 80 pES patients according to genetic subgroups
| M+, N = 52 | M−, N = 28 | P, M+ vs M− | Pathogenic, N = 32 | Probably pathogenic, N = 20 | P, pathogenic vs probably pathogenic | |
|---|---|---|---|---|---|---|
| Consanguinity, % (n) | 16 (9) | 7 (2) | 19 (6) | 15 (3) | ||
| Immune manifestations in first-degree relatives, % (n) | 37 (19) | 18 (5) | .08 | 44 (14) | 25 (5) | |
| Immune manifestations in extended relatives, % (n) | 50 (26) | 30 (8) | .06 | 57 (18) | 38 (8) | |
| Sex ratio (F/M) | 0.7 | 0.6 | 0.8 | 0.7 | ||
| Median age at first episode of cytopenia (min-max), y | 5.2 (0.5-15.2) | 7.1 (0.3-15.6) | 6.8 (0.5-15.2) | 4.1 (0.8-14.7) | ||
| Associated immunopathological manifestations, % (n) | 92 (48) | 64 (18) | .001 | 97 (31) | 85 (17) | |
| Median age at first immunopathologic manifestation (min-max), y | 5.3 (0.3-14.9) | 8.6 (1.6-17.2) | 5.0 (0.3-14.9) | 5.5 (0.8-17.7) | ||
| Median no. of immunopathologic manifestations (min-max) | 2.5 (0-8) | 1 (0-6) | .007 | 3 (0-8) | 2 (0-4) | |
| Median age at immunological disease onset (ES or IM) (min-max), y | 3.3 (0.3-14.7) | 6.0 (0.3-15.6) | 3.9 (0.3-14.1) | 2.6 (0.8-14.7) | ||
| Lymphoproliferation, % (n) | 58 (30) | 35 (10) | .06 | 63 (20) | 50 (10) | |
| Hypogammaglobulinemia, % (n) | 56 (29) | 29 (8) | .02 | 60 (19) | 50 (10) | |
| Autoimmune/autoinflammatory organ disease, % (n) | 56 (29) | 32 (9) | .04 | 63 (20) | 45 (9) | |
| Liver and digestive tract manifestations, % | 13 | 2 | .051 | 12 | 1 | .008 |
| Lung manifestations, % | 10 | 2 | 9 | 1 | .07 | |
| Neurologic manifestations, % | 8 | 2 | 5 | 3 | ||
| Skin manifestations, % | 6 | 4 | 5 | 1 | ||
| Rheumatologic manifestations, % | 3 | 1 | 2 | 1 | ||
| Endocrinologic manifestations, % | 3 | 0 | 2 | 1 | ||
| Atopy, % (n) | 25 (13) | 7 (2) | .07 | 28 (9) | 20 (4) | |
| SLE biomarkers,* % (n) | 17 (9) | 36 (10) | 16 (5) | 20 (4) | ||
| ALPS biomarkers† % (n) | 19 (10) | 7 (3) | 28 (9) | 5 (1) | .07 | |
| Severe or recurrent infections, % (n) | 50 (26) | 43 (12) | 57 (18) | 40 (8) | ||
| Median follow-up from first cytopenia (min-max), y | 9.6 (0.9-25.0) | 7.0 (0.2-26.5) | 9.2 (0.9-25.0) | 10.2 (4.2-20.8) | ||
| Need for second-line treatments, % (n) | 81 (42) | 63 (18) | 88 (28) | 70 (14) | ||
| Median no. of second-line treatments (min-max) | 2 (0-9) | 1 (0-5) | .02 | 2 (0-9) | 2 (0-6) | |
| Deaths, % (n) | 12 (6) | 0 (0) | .086 | 12 (4) | 10 (2) |
| M+, N = 52 | M−, N = 28 | P, M+ vs M− | Pathogenic, N = 32 | Probably pathogenic, N = 20 | P, pathogenic vs probably pathogenic | |
|---|---|---|---|---|---|---|
| Consanguinity, % (n) | 16 (9) | 7 (2) | 19 (6) | 15 (3) | ||
| Immune manifestations in first-degree relatives, % (n) | 37 (19) | 18 (5) | .08 | 44 (14) | 25 (5) | |
| Immune manifestations in extended relatives, % (n) | 50 (26) | 30 (8) | .06 | 57 (18) | 38 (8) | |
| Sex ratio (F/M) | 0.7 | 0.6 | 0.8 | 0.7 | ||
| Median age at first episode of cytopenia (min-max), y | 5.2 (0.5-15.2) | 7.1 (0.3-15.6) | 6.8 (0.5-15.2) | 4.1 (0.8-14.7) | ||
| Associated immunopathological manifestations, % (n) | 92 (48) | 64 (18) | .001 | 97 (31) | 85 (17) | |
| Median age at first immunopathologic manifestation (min-max), y | 5.3 (0.3-14.9) | 8.6 (1.6-17.2) | 5.0 (0.3-14.9) | 5.5 (0.8-17.7) | ||
| Median no. of immunopathologic manifestations (min-max) | 2.5 (0-8) | 1 (0-6) | .007 | 3 (0-8) | 2 (0-4) | |
| Median age at immunological disease onset (ES or IM) (min-max), y | 3.3 (0.3-14.7) | 6.0 (0.3-15.6) | 3.9 (0.3-14.1) | 2.6 (0.8-14.7) | ||
| Lymphoproliferation, % (n) | 58 (30) | 35 (10) | .06 | 63 (20) | 50 (10) | |
| Hypogammaglobulinemia, % (n) | 56 (29) | 29 (8) | .02 | 60 (19) | 50 (10) | |
| Autoimmune/autoinflammatory organ disease, % (n) | 56 (29) | 32 (9) | .04 | 63 (20) | 45 (9) | |
| Liver and digestive tract manifestations, % | 13 | 2 | .051 | 12 | 1 | .008 |
| Lung manifestations, % | 10 | 2 | 9 | 1 | .07 | |
| Neurologic manifestations, % | 8 | 2 | 5 | 3 | ||
| Skin manifestations, % | 6 | 4 | 5 | 1 | ||
| Rheumatologic manifestations, % | 3 | 1 | 2 | 1 | ||
| Endocrinologic manifestations, % | 3 | 0 | 2 | 1 | ||
| Atopy, % (n) | 25 (13) | 7 (2) | .07 | 28 (9) | 20 (4) | |
| SLE biomarkers,* % (n) | 17 (9) | 36 (10) | 16 (5) | 20 (4) | ||
| ALPS biomarkers† % (n) | 19 (10) | 7 (3) | 28 (9) | 5 (1) | .07 | |
| Severe or recurrent infections, % (n) | 50 (26) | 43 (12) | 57 (18) | 40 (8) | ||
| Median follow-up from first cytopenia (min-max), y | 9.6 (0.9-25.0) | 7.0 (0.2-26.5) | 9.2 (0.9-25.0) | 10.2 (4.2-20.8) | ||
| Need for second-line treatments, % (n) | 81 (42) | 63 (18) | 88 (28) | 70 (14) | ||
| Median no. of second-line treatments (min-max) | 2 (0-9) | 1 (0-5) | .02 | 2 (0-9) | 2 (0-6) | |
| Deaths, % (n) | 12 (6) | 0 (0) | .086 | 12 (4) | 10 (2) |
Bold values represent P < .05, considered statistically significant.
ALPS, autoimmune lymphoproliferative syndrome; F, female; IM, immunopathological manifestation; M, male; M+, patients with mutations; M−, patients without detected mutations; SLE, systemic lupus erythematosus.
SLE biomarkers: antinuclear antibodies titer >1/160 on 2 separate samples, isolated significant autoantibodies.
ALPS biomarkers: persistent hypergammaglobulinemia (at least 2 SD over the mean for age), high counts of circulating TCR ab CD4−CD8− double-negative T lymphocytes.
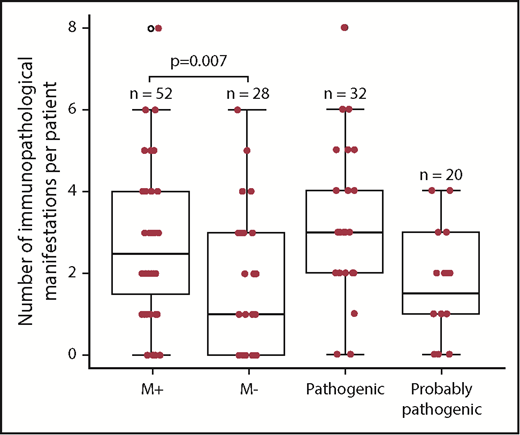
A box plot representation of the number of immunopathologic manifestations per patient, as a function of the genetic subgroup in 80 pES patients (the M+, M−, “pathogenic” and “probably pathogenic” groups). The bottom of the box marks the 25th percentile, the median line marks the 50th percentile, and the top of the box marks the 75th percentile. The bottom of the vertical line indicates the minimum and the maximum. The vertical lines at the top and bottom indicate the minimum and the maximum. The symbols at the top and bottom indicate outlying data points.
A box plot representation of the number of immunopathologic manifestations per patient, as a function of the genetic subgroup in 80 pES patients (the M+, M−, “pathogenic” and “probably pathogenic” groups). The bottom of the box marks the 25th percentile, the median line marks the 50th percentile, and the top of the box marks the 75th percentile. The bottom of the vertical line indicates the minimum and the maximum. The vertical lines at the top and bottom indicate the minimum and the maximum. The symbols at the top and bottom indicate outlying data points.

Patients with pES free of additional immunopathologic manifestations, as a function of the genetic subgroup.
Patients with pES free of additional immunopathologic manifestations, as a function of the genetic subgroup.
Discussion
The results of the present prospective study showed that pathogenic or probably pathogenic genetic variants (most of which were germline mutations) can be detected in the majority of children with ES. The generation of these novel data was made possible by accessing a large national prospective cohort of patients with pES and NGS resources.44,45 It was already known that children with monogenic inherited disorders of the immune system (ie, PIDs) are 80- to 120-fold more likely to develop chronic immune cytopenia than an age-matched population is.3,46,47 In adult patients, Michel et al48 have shown that ES was a secondary condition in 56% of cases (mainly as a results of malignancies and autoimmune diseases); a PID was identified in only 9% of cases. Given the estimated prevalence of PIDs, it has been predicted that at least one-third of children with chronic immune cytopenia have a causal genetic disorder.3 We therefore decided to perform a genetic analysis in a large group of patients from the French OBS’CEREVANCE pES cohort. We did not apply any a priori selection criteria other than consent by the family and/or patient. The study group was similar (but not identical) to the overall pES cohort. Our results show that in 65% of the patients, pES was strongly associated with the detection of significant genetic variants. These results will be addressed for each of 3 observed groups, that is, patients with known genetic variants, patients with new potentially pathogenic genetic variants, and patients with no detected pathogenic or probably pathogenic genetic variants.
Unsurprisingly, a number of patients carried mutations known to be strong risk factors for autoimmune cytopenia. These included germline and somatic TNFRSF6 mutations causing ALPS,4,49 mutations in the CTLA4 and LRBA genes,16,20 GOF mutations in STAT350 or PiK3CD,17 hypomorphic LOF mutations in RAG1,51 and somatic mutations in CBL and KRAS associated with RALD.52,53 Despite the presence of associated clinical manifestations in some of the patients, it is noteworthy that none of them (with the exception of cases of ALPS) had previously received a firm diagnosis. Although autoimmune cytopenia is not uncommon in these conditions, it is not always present.16-18,20,27,50-55 It is also known that some TNFRSF6 and CTLA4 mutations do not have full penetrance.18,54 Hence, additional predisposing factors (possibly environmental and/or genetic factors) may favor the occurrence of ES in these children.1,2 In this respect, the high overall heritability of the genomic variations in many pediatric autoimmune conditions (although not in autoimmune cytopenia) identified in genome-wide association studies might represent a “favorable” background on which a critical gene variant (CTLA4, LRBA) causes autoimmunity.2 The high proportion of patients with a family history of autoimmune disease (42% when considering first-degree relatives, and 57% when considering extended families, values that are greater than expected for diseases with autosomal-dominant inheritance) fits with this view. Based on these results, performance of the same genetic analysis in the patients’ relatives is an obvious next step.
The putative causal role of the probably pathogenic variants identified in the second (“probably pathogenic”) group of patients must be scrutinized with caution. Strict criteria were used (in terms of both allele frequency and predicted pathogenicity) to select the detected variants. The gene product’s function (if known from the literature) was also considered. It is noteworthy that variants in 5 genes were found in unrelated families, further suggesting causality. Other variants in some of these genes are known to be pathogenic in contexts other than autoimmunity.28,29,39,56 The observation that the phenotypic characteristics of this patient group were very similar to those of patients carrying known pathogenic variants also argues in favor of the variants’ pathogenicity. Relative to the M− group, patients in the M+ group were more likely to have more additional immunopathologic events. Twenty-five patients developed hypogammaglobulinemia after rituximab treatment, as previously reported.57 Still, given its efficacy, rituximab should probably be considered as the first-line treatment in pES. However, a careful evaluation is warranted before rituximab initiation and during follow-up. All of the associated immunopathologic events occurred earlier in the M+ group, required more frequent immunosuppressive therapy, and tended to be associated with a poorer prognosis (ie, poorer survival). However, it must be noted there were some phenotypic differences between patients with known mutations and patients with probably pathogenic variants; the latter group were slightly (but not significantly) less likely to have a family history of immunopathology, a personal history of digestive tract disease and/or ALPS biomarkers. The latter findings may be explained, respectively, by (i) the presence of a number of ALPS patients in the first group, and (ii) the presence of cases at risk of inflammatory bowel disease in the same group (ie, LRBA and CTLA4 deficiencies, and STAT3 GOF mutations).18,20,27 We therefore hypothesize that the “probably pathogenic” variants detected here (see below) are risk factors that are mostly (but not exclusively) related to the control or occurrence of antibody-mediated autoimmunity.
The detection of “probably pathogenic” variants may broaden the field of genetic predisposition to autoimmunity. Several mechanisms can be considered: (i) impaired suppression of conventional T cell proliferation by regulatory T cells,58-60 (ii) GOF or LOF variants that increase immune effector function (in various ways) downstream of cytokines and antigen-specific B- or T-cell receptors or co-receptors, or (iii) pathologic changes in immune responses in the context of infection or the control of skin and gut microbiotas.28,32,39,61-69 A fourth potential mechanism relates to lymphocyte apoptosis, which is likely to be defective in patients with RIPK2 and APAF1 mutations.70,71 All of these variants must now undergo extensive functional validation, in order to characterize the mechanism predisposing to autoimmunity. It will probably also be of value to extend screening for the variants described here to other children with ES and to pediatric cohorts with other autoimmune diseases.
The present results do not imply that the known genetic variants (within the M+ group) caused an autoimmune disease with monogenic Mendelian inheritance. The familial segregation of these mutations will now also have to be studied. It is important to bear in mind that no other significant immune gene variants were detected in these patients. Nevertheless, this observation does not rule out a role of other gene variants in general and regulatory variants (expressed quantitative trait loci) in particular.72
Lastly, genetic factors may be less critical in the third group of patients with pES, that is, those in whom no significant genetic variants were found with our genetic testing strategy. Overall, this group of patients is distinct from the other 2 groups because its members were less likely to present with additional immunopathologic features (including allergy) and had somewhat less severe ES. Nevertheless, a family history of autoimmunity was not uncommon, suggesting that polygenic genetic variations are involved, given the high heritability of autoimmunity in children.2
Our genetic approach had some limitations, and may have failed to detect variants in some cases (ie, false-negatives). Most of the patients were screened using tNGS; we cannot rule out the possibility that mutations in genes not included in the panel, intronic variants or somatic mutations of known genes not detected by the present testing strategy could be involved in the M− group. Further analyses (such as whole-genome sequencing or the in-depth sequencing of candidate genes in lymphocyte subsets73 ) should therefore be performed before a genetic cause is ruled out.
In conclusion, our results suggest that wide-ranging genetic screening should be offered to children with ES because the findings have prognostic significance and may thus influence treatment choices. Indeed, we identified 29 patients (36%) with mutations that might help the physician to choose a targeted treatment of severe autoimmune cytopenia, as shown (for instance) by the use of (i) a mechanistic target or rapamycin inhibitors in patients with ALPS74 or an activated phosphoinositide 3-kinase delta (PIK3δ) syndrome,75 (ii) CTLA-4 fusion protein therapy in CTLA-4– and LRBA-deficient patients,18,20 (iii) the potential use of JAK inhibitors to targeting GOF variants in JAK1 or JAK2,39 and possibly (iv) calcineurin inhibitor therapy in patients with NFATC1 variants.76 The present study raises new questions about the genetic background of pES and, more widely, autoimmune diseases in children. It is now justifiable to extend genetic testing to all children with chronic multilineage cytopenia, their family members, and other cohorts of pediatric patients with autoimmune diseases. Our study also provides a basis for gaining further insights into the mechanistic aspects of controlling reactivity to self, along with the many checkpoints already known.77,78 We expect this type of study to provide additional insight into the pathophysiology of autoimmunity, together with important patient-level information with prognostic and therapeutic value.
For original data, please contact frederic.rieux-laucat@inserm.fr.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank all of the medical teams involved in the CEREVANCE prospective cohort study from 2004 onward; please note that the numbers of patients with pES included are shown in parentheses (total, n = 203): G. Leverger (25), S. Ducassou (23), B. Neven (19), Y. Bertrand (15), V. Barlogis (14), M. Pasquet (13), W. Abou Chahla (12), T. Leblanc (10), C. Guitton (7), I. Pellier (6), E. Jeziorski (6), F. Monpoux (6), P. Blouin (6), C. Armari-Alla (5), F. Fouyssac (5), C. Thomas (5), E. Colomb Bottollier (4), V. Gandemer (4), A. Marie-Cardine (4), C. Paillard (4), F. Millot (3), N. Cheikh (2), C. Piguet (2), L. Carausou (1), M. Deparis (1), E. Dore (1).
This work was supported by the Institut des Maladies Rares (GIS; INSERM), the French Ministry of Health (Rare Disease Plan, Programme Hospitalier de Recherche Clinique [PHRC] 2005), the Association Bordelaise pour l’Avancement des Sciences Pédiatriques (ABASP) research charity, the Association pour la Recherche et les Maladies Hématologiques de l’Enfant (RMHE) research charity, and the Association Française de Science Economique (AFSE) and O-CYTO patient associations.
Authorship
Contribution: F.R.-L., A.F., and B.N. designed the study; J.H., N.A., H.F., and F.R.-L. acquired the data; J.H., N.A., H.F., B.N., A.F., and F.R.-L. drafted the manuscript; and all of the authors analyzed and interpreted the data and revised the manuscript for critical content.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
A complete list of the members of the French Reference Center For Pediatric Autoimmune Cytopenia (CEREVANCE) appears in the supplemental Appendix.
Correspondence: Frederic Rieux-Laucat, Laboratory of Immunogenetics of Pediatric Autoimmune Diseases, INSERM UMR 1163-Institut Imagine, 24 Blvd du Montparnasse, F-75015, Paris, France; e-mail: frederic.rieux-laucat@inserm.fr.
