

TO THE EDITOR:
B-cell precursor acute lymphoblastic leukemia (BCP-ALL) is the most common childhood cancer and is molecularly heterogeneous.1 Identical twins with concordant BCP-ALL have provided a unique and tractable model to delineate the natural history and clonal evolution of the disease.1-3 Such studies have led to the 2-hit model for pediatric BCP-ALL, elucidated by Greaves,4 which provides unambiguous evidence that an initiating alteration, often occurring in utero, generates a preleukemic clone, which eventually gives rise to an overt leukemia upon acquisition of secondary cooperating mutations.5 Importantly, genome-wide sequencing has revealed a strikingly silent mutational landscape in childhood BCP-ALL, further indicative of a developmental cancer of prenatal origin, with a short window of time to accumulate molecular alterations.6-8
The natural history of the disease has been studied in major cytogenetic subgroups of childhood BCP-ALL, including MLL fusion,9-11 ETV6-RUNX1+,12-14 hyperdiploid,3,5,15,16 BCR-ABL1+, and Ikaros mutated.17 However, formal demonstration of the prenatal origin and backtracking of the natural history of the leukemia remain understudied in the major subgroup, so-called B-other ALL. This subgroup accounts for ∼30% of de novo diagnoses and is defined by the absence of main classifying alterations (ie, chromosomal translocations, aneuploidy, and copy-number alterations and/or mutations recurrently observed in BCP-ALL), thus challenging the use of patient-specific molecular tags to backtrack the natural history of the disease.1,18-21
Predicated based on the hypothesis that shared identical mutations are prenatal in origin and twin-specific mutations are likely to be postnatal and secondary, we here performed a genome-wide multilayer OMICs analysis, including whole-genome DNA (WG-seq), B-cell receptor (BCR-seq), and whole-genome DNA bisulphite sequencing (WGB-seq) on a pair of 8-month-old monozygotic twins diagnosed with concordant B-other BCP-ALL. We demonstrate a parallel and convergent genetic and epigenetic evolution of a TCF3-ZNF384/PTPN11–driven concordant B-other BCP-ALL and support a pre-VDJ primitive fetal hematopoietic progenitor or stem cell as the cell of origin of TCF3-ZNF384 and PTPN11 mutations.
Routine molecular diagnostics of monozygotic twins with concordant BCP-ALL revealed the absence of molecular/cytogenetic alterations associated with BCP-ALL (supplemental Table 1, available on the Blood Web site; supplemental Figure 1), thus classifying the leukemia as B-other BCP-ALL.8,20 We therefore performed high-coverage WG-seq in purified blasts and peripheral blood cells at complete remission from both twins to comprehensively characterize the developmental timing of acquired somatic mutations (WG-seq detailed in supplemental Table 2). We found a total of 227 and 261 somatic mutations in twin A and twin B, respectively (Figure 1A; supplemental Table 3), which represents a low mutational load (∼0.07 mutations per Mb) and is in line with the silent mutational landscape reported for infant BCP-ALL.6,7 Notably, 75 somatic mutations were shared between both twins, suggesting a common prenatal origin of the leukemia (Figure 1A-B). Only 3 of these mutations affected coding regions: t(12;19)(p13;p13) encoding the TCF3-ZNF384 fusion gene (Figure 1A; supplemental Figure 2A)22 and point mutations in SDAD1 and UQCR11 (Figure 1B). These prenatal coding mutations showed a mutant allele frequency nearing 50%, thus representing initial events in the original leukemic clone. The t(12;19)(p13;p13)/TCF3-ZNF384 rearrangement showed an identical genomic breakpoint in both twins (supplemental Figure 2A) and was confirmed by fluorescence in situ hybridization (supplemental Figure 2B) and RT-PCR (supplemental Figure 2C) and sequence validated (supplemental Figure 2D). The other coding mutations were validated using orthogonal sequencing strategies (supplemental Table 4).
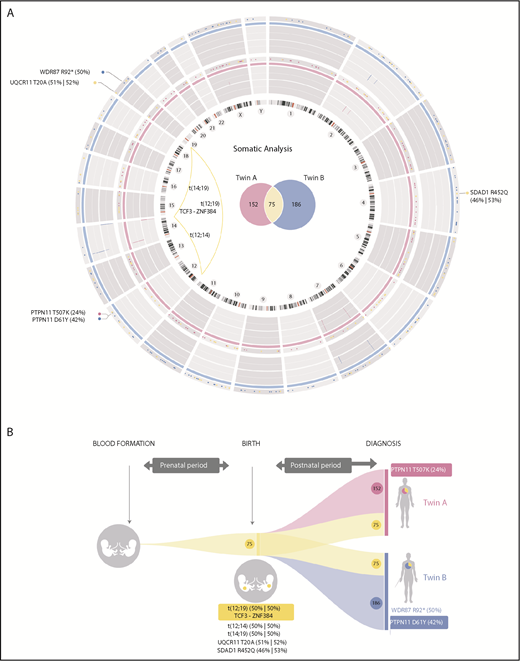
Mutational landscape and natural history reconstruction of BCP-ALL monozygotic twins. (A) Circos plot (http://circos.ca/) representing the total number of mutations identified at diagnosis for each twin. Twin A is shown in pink (inner ring). Twin B is shown in blue (outer ring). Chromosome ideograms are indicated inside the graph. Copy-number alterations are represented as lines within the gray rings. Venn diagrams inside the graph show the total number of exclusive (pink, twin A; blue, twin B) and shared (yellow) somatic mutations. Coding mutations are annotated outside the graph according to genomic gene location. Substitution mutant allele frequency is shown in brackets. Genomic chromosomal rearrangements are represented with yellow lines connecting chromosomal breakpoints. (B) Graphical reconstruction of the number of mutations accumulated during tumor history. Approximately one-third of the somatic mutations (shown in yellow), including the translocation responsible for the expression of the TCF3-ZNF384 fusion gene, occurred before birth, when both twins shared the same circulatory system. Coding mutations are indicated. Leukemia in both twins evolved independently after birth, with twin-specific postnatal mutations.
Mutational landscape and natural history reconstruction of BCP-ALL monozygotic twins. (A) Circos plot (http://circos.ca/) representing the total number of mutations identified at diagnosis for each twin. Twin A is shown in pink (inner ring). Twin B is shown in blue (outer ring). Chromosome ideograms are indicated inside the graph. Copy-number alterations are represented as lines within the gray rings. Venn diagrams inside the graph show the total number of exclusive (pink, twin A; blue, twin B) and shared (yellow) somatic mutations. Coding mutations are annotated outside the graph according to genomic gene location. Substitution mutant allele frequency is shown in brackets. Genomic chromosomal rearrangements are represented with yellow lines connecting chromosomal breakpoints. (B) Graphical reconstruction of the number of mutations accumulated during tumor history. Approximately one-third of the somatic mutations (shown in yellow), including the translocation responsible for the expression of the TCF3-ZNF384 fusion gene, occurred before birth, when both twins shared the same circulatory system. Coding mutations are indicated. Leukemia in both twins evolved independently after birth, with twin-specific postnatal mutations.
We additionally found 152 and 186 somatic mutations exclusive to twin A and twin B, respectively, suggesting that, during individual development, the leukemias evolved in parallel, and twin-specific mutations accumulated postnatally (Figure 1A-B; supplemental Table 3). Regarding coding mutations, each twin acquired a distinct nonsynonymous mutation in PTPN1123 (Figure 1A-B; supplemental Figure 3). Both coding mutations in PTPN11 were subclonal and predicted by SHIFT and POLYPHEN algorithms to affect evolutionary conserved amino acids and significantly affect the function of the protein domains involved. The 2 identified mutations (D61Y and T507K) affect domains that modulate PTPN11 phosphatase activity, previously described to increase PTPN11 activity. Therefore, these PTPN11 mutations likely represent gain-of-function postnatal secondary oncogenic hits, promoting proliferation and cooperating with TCF3-ZNF384 for overt leukemia development, suggesting a convergent evolution in both leukemias.
To further explore the evolutionary traits of the concordant BCP-ALL in both twins, we used WGB-seq to survey the DNA methylation status of concordant BCP-ALLs (WGB-seq detailed in supplemental Table 5). BCP-ALL cells from both twins were significantly hypomethylated as compared with their healthy FL-BCP counterparts (Figure 2A). In fact, among the ∼190 000 and ∼176 000 dmCpG sites found for twin A and B, respectively, the number of hypomethylated CpG sites largely outperformed the number of hypermethylated sites (Figure 2B). However, we observed limited DNA methylation changes between the twins’ leukemic cells (Figure 2B green bars). Indeed, both twins displayed a similar enrichment of dmCpG sites, both hyper- and hypomethylated, with regard to their CpG context (Figure 2C) or CpG genomic location (Figure 2C-E). The relatively few dmCpG sites observed between twins might reflect interindividual variability rather than a divergent epigenetic evolution of the leukemia, reinforcing a parallel and convergent genetic, but also epigenetic, evolution of the BCP-ALL in this pair of monozygotic twins.
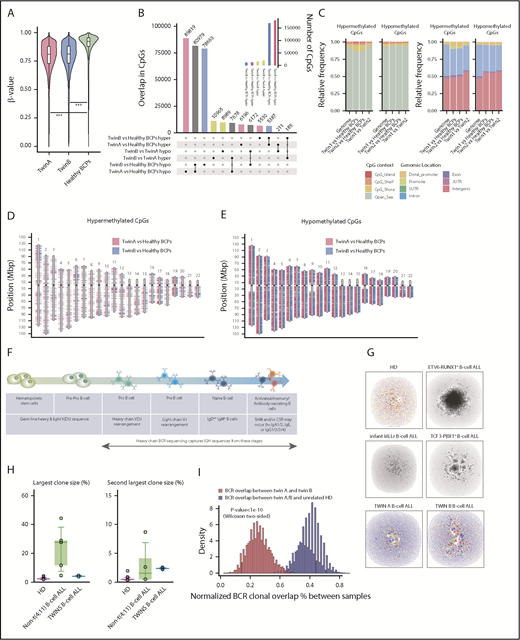
Genome-wide DNA methylation analysis and BCR repertoires in BCP-ALL monozygotic twins. (A) Violin plots depicting the global DNA methylation levels (β-value distributions) of CpG sites identified by WGB-seq. For healthy BCPs, the average DNA methylation value of 2 FL-derived pools of BCP is represented. (B) Total number of differentially hyper- (hyper) and hypomethylated (hypo) CpGs observed for the indicated comparisons between twins and healthy BCPs. The inner graph represents the total number of differentially methylated CpG (dmCpG) sites. (C) Stacked bar plots indicating the relative proportion of significant hyper- and hypomethylated CpGs between twins, or between each twin and healthy BCPs, regarding their CpG context (left) or the CpG genomic location (right), respectively, as indicated in the colored legend. (D-E) Ideograms representing the genomic location of differentially DNA hypermethylated (D) and hypomethylated (E) CpG sites obtained for twin A (pink) and twin B (blue) as compared with healthy BCPs. (F) Cartoon summarizing how BCR-seq captures IgH sequences across B-cell stages/development. (G) Network plots of BCR repertoires of a representative healthy donor (HD) and an infant mixed-lineage leukemia–rearranged (rMLL) pro-B ALL depicting the existence of many minor diagnostic nonexpanded B-cell clones (top middle left). Network plots of BCR repertoires of a representative t(12;21)/ETV6-RUNX1+ and t(1;19)/TCF3-PBX1+ pediatric BCP-ALL showing high clonality of B-cell clones (top middle right). Network plots of BCR repertoires of TCF3-ZNF384–rearranged pro-B ALL in both monozygotic twins (bottom). Each vertex represents a unique BCR sequence, and relative vertex size is proportional to the number of identical reads. For the HD and BCP-ALL twin samples, each vertex is represented by a pie chart indicating the percentage of each isotype, where blue = IgD/M, red = IgA1/2, yellow = IgG1/2, green = IgG3, and gray = IgE. (H) Boxplots of the largest and second largest BCR clone sizes for HD, non-t(4;11)+ BCP-ALL, and twins with concordant BCP-ALL. (I) Histogram representing the number of BCR clones shared between twin A and twin B (blue) or between unrelated HD and the twins (red).
Genome-wide DNA methylation analysis and BCR repertoires in BCP-ALL monozygotic twins. (A) Violin plots depicting the global DNA methylation levels (β-value distributions) of CpG sites identified by WGB-seq. For healthy BCPs, the average DNA methylation value of 2 FL-derived pools of BCP is represented. (B) Total number of differentially hyper- (hyper) and hypomethylated (hypo) CpGs observed for the indicated comparisons between twins and healthy BCPs. The inner graph represents the total number of differentially methylated CpG (dmCpG) sites. (C) Stacked bar plots indicating the relative proportion of significant hyper- and hypomethylated CpGs between twins, or between each twin and healthy BCPs, regarding their CpG context (left) or the CpG genomic location (right), respectively, as indicated in the colored legend. (D-E) Ideograms representing the genomic location of differentially DNA hypermethylated (D) and hypomethylated (E) CpG sites obtained for twin A (pink) and twin B (blue) as compared with healthy BCPs. (F) Cartoon summarizing how BCR-seq captures IgH sequences across B-cell stages/development. (G) Network plots of BCR repertoires of a representative healthy donor (HD) and an infant mixed-lineage leukemia–rearranged (rMLL) pro-B ALL depicting the existence of many minor diagnostic nonexpanded B-cell clones (top middle left). Network plots of BCR repertoires of a representative t(12;21)/ETV6-RUNX1+ and t(1;19)/TCF3-PBX1+ pediatric BCP-ALL showing high clonality of B-cell clones (top middle right). Network plots of BCR repertoires of TCF3-ZNF384–rearranged pro-B ALL in both monozygotic twins (bottom). Each vertex represents a unique BCR sequence, and relative vertex size is proportional to the number of identical reads. For the HD and BCP-ALL twin samples, each vertex is represented by a pie chart indicating the percentage of each isotype, where blue = IgD/M, red = IgA1/2, yellow = IgG1/2, green = IgG3, and gray = IgE. (H) Boxplots of the largest and second largest BCR clone sizes for HD, non-t(4;11)+ BCP-ALL, and twins with concordant BCP-ALL. (I) Histogram representing the number of BCR clones shared between twin A and twin B (blue) or between unrelated HD and the twins (red).
Whether pathogenic chromosomal rearrangements and/or protein-coding DNA mutations in cancer are associated with chromatin and/or DNA methylation changes remains elusive. We therefore wanted to gain insight into the methylation status of the loci specifically found rearranged or mutated in both twins (TCF3, ZNF384, SDAD1, UQCR11, WRD87, and PTPN11). Strikingly, in contrast to healthy FL-BCPs and BCP-ALL cells from unrelated infant B-other ALLs, we found that BCP-ALL cells from both twins displayed concordant and specific significant DNA hypomethylation in TCF3 and ZNF384 loci, in the vicinity of the translocation genomic breakpoints (supplemental Figure 4A-B), but also within the promoter or body gene region of SDAD1, WDR87, and PTPN11, coinciding with CpG islands and shore regions (supplemental Figure 4C-F). Importantly, DNA methylation changes were not observed in MLL or TP53 loci (supplemental Figure 4G-H), supporting the specificity of the DNA hypomethylation observed in the loci rearranged/mutated in leukemic twins.
We next analyzed BCR repertoires to study the clonal composition of immunoglobulin heavy chain (IgH) rearrangements in diagnostic samples from both twins. BCRs are generated during B-cell differentiation (Figure 2F) and represent unique molecular tags for each B-cell clone through the recombination of V, D, and J genes, thus providing a detailed view of the B-cell population dynamics and clone tracking.6,24 We therefore performed BCR-seq on TCF3-ZNF384+ BCP-ALL cells from both twins to address whether they expressed fully rearranged BCRs, from which major B-cell clonal expansion may be observed.6,24 After BCR-seq filtering, each sample yielded 6362 and 11 832 unique BCRs for twin A and B, respectively. Similar to that reported for infant mixed-lineage leukemia–rearranged pro-B ALL, BCR repertoires from infant TCF3-ZNF384+ BCP-ALL twins did not exhibit significantly expanded VDJ-rearranged B-cell clones (Figure 2G-H). This is in contrast to the significantly large B-cell clones consistently observed in noninfant t(1;19)/TCF3-PBX1+, t(12;21)/ETV6-RUNX1+, and t(9;22)/BCR-ABL1+ BCP-ALLs.
Because the TCF3-ZNF384, SDAD1, and UQCR11 mutations are clonal, and PTPN11 mutations are found in major clones, the lack of B-cell clonal expansion based on IgH rearrangements, together with the leukemia phenotype developmentally stalled at the pro-B stage, supports a model in which the genomic drivers in this pair of infant leukemic twins originate in a primitive fetal progenitor with a germ line or an incompletely rearranged (pre-VDJ) IgH locus.6 The fifth Ewing variant transcription factor is uniquely expressed in fetal primitive hematopoietic stem and progenitor cells and has been shown to be expressed on a vast majority of infant leukemias with a prenatal origin.25 Through a diagnostic approach, we confirmed a significantly higher expression of fifth Ewing variant transcription factor in both leukemic twins and infant/pediatric B-ALLs (n = 11) than in adult B-ALLs (n = 5), further supporting the primitive fetal progenitor nature of the cell of origin in both leukemic twins (supplemental Figure 5). Given that IgH rearrangement is a highly epigenetically regulated process, we would predict a higher similarity of VDJ rearrangements between the twins, given their shared mutations and epigenetic landscapes. Indeed, the BCR clonal overlap between the twins was threefold higher than that between twins and healthy donors (Figure 2I). The shared IgH sequences between the twins comprised multiple small clones, primarily IgD/M, with a strong concordance of isotype usage between the twins (supplemental Figure 6). Together, these data demonstrate the concordance between B-cell populations in both infant twins, consistent with shared epigenetic landscapes of B-cell progenitor populations.
Finally, bone marrow–derived mesenchymal stem cells (BM-MSCs) play a role in the pathogenesis of a variety of hematological malignances,26 and leukemia-specific genetic alterations have been found in BM-MSCs from MLL-rearranged infant BCP-ALL patients, indicating that stromal cells may be in part tumor related and that such oncogenic insults may arise in a population of prehematopoietic precursors.27,28 To further ascertain the cellular origin of the coding mutations found in our leukemic twins, we generated and characterized bona fide BM-MSCs cultures29 and used fluorescence in situ hybridization and targeted sequencing to search for the presence of both shared and twin-specific coding mutations found in the leukemic cells. Such coding mutations were not detected in BM-MSCs from either twin, suggesting that BM-MSCs do not seem to be tumor related in these leukemic twins. This further confirms that both shared and twin-specific coding mutations arise in an early hematopoietic fetal pre-VDJ progenitor or stem cell, rather than in prehematopoietic precursors, and provides insight into the timing of these mutations. In sum, this is the first report on the natural history, evolutionary trajectories, and cell of origin of TCF3-ZNF384 and PTPN11 mutations in a pair of infant monozygotic twins with concordant B-other BCP-ALL. WG-seq and WGB-seq data sets have been deposited in the European Genome-Phenome Archive under the accession numbers EGAD00001005017 and EGAD00001005018, respectively. Data access will be granted upon request.
The online version of this article contains a data supplement.
Acknowledgments
The authors thank Mireia Camos (Hospital Sant Joan de Déu Barcelona, Barcelona, Spain) for critical reading of the manuscript.
This work was supported by European Research Council grants CoG-2014-646903 and PoC-2018-811220 (P.M.) and StG-2014-637904 (I.V.), Generalitat de Catalunya grants SGR330 and PERIS 2017-2019 (P.M.), Spanish Ministry of Economy and Competitiveness grants SAF2016-80481-R and SAF2016-76758-R (P.M. and I.V.), Spanish Association Against Cancer grant AECC-CI-2015 (C.B.), Health Institute Carlos III grant PI17/01028 (C.B.), and a FERO Foundation grant (C.B.). P.M. also acknowledges financial support from the Obra Social La Caixa-Fundació Josep Carreras and Fundación Leo Messi. J.R.T. is supported by a Juan de la Cierva Formación fellowship from the Spanish Ministry of Science and Innovation (FJCI-2015-26965) and Institute of Oncology of Asturias. R.V.-M. is supported by a Torres Quevedo fellowship from the Spanish Ministry of Science and Innovation (PTQ-16-08623). P.M. is an investigator of the Spanish Cell Therapy Cooperative Network.
Authorship
Contribution: C.B., J.R.T., R.B.-R., L.G.-S., R.V.-M., A.A.-D., R.D.d.l.G., J.R., and L.Z. performed experiments and analyzed data; C.B., A.F.F., M.F.F., C.B.-N., N.A., H.G., E.G., G.L., P.B., I.V., and P.M. conceived the study, designed experiments, and analyzed data; and C.B., J.R.T., R.B.-R., R.V.-M., L.G.-S., I.V., and P.M. contributed to artwork and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Paola Ballerini, Service d’Hématologie Biologique, Hospital Armand Trousseau, Paris, France; e-mail: paola.ballerini@aphp.fr; Ignacio Varela, Instituto de Biomedicina y Biotecnología de Cantabria, Universidad de Cantabria–CSIC, Santander, Spain; e-mail: ignacio.varela@unican.es; and Pablo Menendez, Josep Carreras Leukemia Research Institute, School of Medicine, University of Barcelona, Casanova 143, 08036 Barcelona, Spain; e-mail: pmenendez@carrerasresearch.org.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal