

Key Points
Venous stasis activates fibrinolysis, which is ineffective because of suppression by α2-antiplasmin.
Lack of α2-antiplasmin activity prevents thrombosis, suggesting that it is essential for venous thrombus formation.

Abstract
Stasis of venous blood triggers deep vein thrombosis by activating coagulation, yet its effects on the fibrinolytic system are not fully understood. We examined the relationship between stasis, fibrinolysis, and the development of experimental venous thrombosis. Effects of stasis-induced deep vein thrombosis and fibrinolysis on thrombosis were examined by inferior vena cava ligation in congenic mice with and without α2-antiplasmin (α2AP), the primary inhibitor of plasmin. Venous thrombus weights were measured and thrombus composition was determined by Martius scarlet blue and immunofluorescence staining. Venous thrombi from α2AP+/+ mice contained plasminogen activators, plasminogen activator inhibitor-1, plasminogen, and α2AP, which changed with thrombus age. Normal, α2AP+/+ mice developed large, occlusive thrombi within 5 hours after ligation; thrombi were even larger in plasminogen-deficient mice (P < .001). No significant thrombus formation was seen in α2AP−/− mice (P < .0001) or in α2AP+/+ mice treated with an α2AP-inactivating antibody (P < .001). Venous stasis activated fibrinolysis, measured by D-dimer levels, in α2AP−/− mice vs α2AP+/+ mice (P < .05). Inhibition of fibrinolysis by the indirect plasmin inhibitor ε-aminocaproic acid or by α2AP restored thrombosis in α2AP−/− mice. In addition to its effects on acute thrombosis, thrombus formation was also markedly suppressed in α2AP−/− mice vs α2AP+/+ mice (P < .0001) 1, 7, and 14 days after ligation. We conclude that experimental venous stasis activates the fibrinolytic system to block the development of venous thrombosis. Suppression of fibrinolysis by α2AP appears essential for stasis-induced thrombus development, which suggests that targeting α2AP may prove useful for preventing venous thrombosis.
Introduction
Venous thromboembolism is the third leading cause of cardiovascular deaths worldwide.1 Postthrombotic syndrome, recurrent thrombosis, and life-threatening pulmonary embolism are serious consequences of deep vein thrombosis.2,3 Since the initial observations of Virchow, blood stasis, hypercoagulability, and vessel wall (endothelial) dysfunction have been identified as triggers of venous thrombosis.1 Tissue factor expression, leukocytes, platelets, and factors XI and XII contribute to the initiation of venous thrombosis.1,4,,-7 In contrast, the endogenous fibrinolytic system is thought to function primarily in the dissolution of already formed, chronic thrombi, and its role in the initial, dynamic phase of thrombus formation has not been established.
Venous thrombosis is triggered by blood stasis, which is a key historical risk factor in patients.1,8 Venous thrombosis occurs over hours following venous occlusion and has been linked to cellular hypoxia9,10 that leads to the formation of thrombi rich in fibrin, platelets, neutrophils, and red blood cells.6,11,12 Prolonged venous stasis has been associated with early reductions in plasminogen activator activity.13 Expression of tissue-type plasminogen activator (tPA) and urinary-type plasminogen activator (uPA) relative to their inhibitor, plasminogen activator inhibitor-1 (PAI-1), is very low in experimental venous thrombi in the first week and increases marginally through 14 days.14 Evidence suggests that uPA contributes to the resolution of acute and chronic venous thrombi,15,16 but little is known about the role of the fibrinolytic system in the stages of early thrombus formation.
In addition to plasminogen activators, fibrinolysis is regulated by a number of inhibitors including α2-antiplasmin (α2AP), a rapid covalent inhibitor of plasmin.17,18 Epidemiologic studies have associated higher blood levels of α2AP with a mild increase in the risk of venous thrombosis.19 Still, there is little information about the role of α2AP or fibrinolysis in the initial generation of venous stasis thrombosis. In this study, we examined the role of venous stasis in activating endogenous fibrinolysis and modulating thrombus formation in a well-established model of venous stasis induced by ligation of the inferior vena cava (IVC).20,21
Materials and methods
Proteins and reagents
Reagents were obtained from the following sources: human α2AP (Athens Research and Technology, Athens, GA); ε-aminocaproic acid (EACA) (Sigma, St. Louis, MO); inhibitory mouse monoclonal antibody against mouse α2AP (#MAP4H9, Molecular Innovations, Novi, MI); and inhibitory monoclonal antibody against human α2AP (TS23, Translational Sciences, Memphis, TN); all the other reagents if not specified (Sigma).
Mouse model of stasis thrombosis
Animal studies were approved by the Institutional Animal Care and Use Committee at University of Tennessee Health Science Center, Memphis, TN, and The University of Arizona, College of Medicine, Phoenix, AZ. Adult male and female C57Black/6J mice and congenic α2AP−/− (KOMP, University of California, Davis, CA) mice were used. Plasminogen (Pg−/−) mice (Jackson Labs, Bar Harbor, ME) were also used. Venous stasis thrombosis was induced by the IVC ligation model as described by Diaz et al.20,21 Mice were anesthetized with 4% isoflurane + 100% oxygen, and continuous anesthesia was given with a mixture of (1.5% to 2%) isoflurane and oxygen (0.2 L/min) during the surgery. For laparotomy, a ventral midline incision was made exposing the inner abdomen. The IVC was carefully exposed and all the side branches and the back branches of the IVC were ligated with nylon 7-0 suture (Ethilon). During our standardization experiments, we found that the back branches of IVC were thick in diameter, similar to the observations of Diaz et al.20 All the back branches were also ligated with nylon 7-0 sutures as a modification of the previous model by Diaz et al.20 The IVC was separated from the aorta, caudal to the left renal vein and ligated with a 7-0 nylon suture.16,20,22 The laparotomy site was closed in a 2-layer fashion. Buprenorphine (50 µg/kg) was administered at the end of surgery to relieve discomfort and the mice were allowed to recover. At different time points after IVC ligation (5 hours, 24 hours, 7 days, 14 days), the mice were euthanized by CO2 asphyxiation, the blood was collected from the heart for plasma preparation in 3.8% sodium citrate (1:9 volume), and whole body perfusion was gently performed through the heart with 15 to 20 mL of 0.9% saline. The IVC tissue, including the thrombus, was harvested from the ligation site (renal branches) to the iliac bifurcation and weighed. IVC tissues from the control animals, who underwent the same surgery without IVC ligation, were used for comparison.
IVC stenosis model of deep vein thrombosis
IVC stenosis was performed using the procedures described in "Mouse model of stasis thrombosis," except that the IVC was ligated over a 30-g blunt needle, which was gently pulled off immediately after ligation to restore partial blood flow, as described by Brill et al.23 The side branches were ligated with a 7-0 nylon suture and the back branches were left open.
A total of 157 mice including α2AP+/+ (N = 70), α2AP−/− (N = 76), and Pg−/− (N = 11) were used. Eight mice (5%) died postsurgery after successful IVC ligation. Mice that died before the IVC harvest time or from surgical errors (eg, rupture of IVC or bleeding in the side or back branches) were not included in the thrombus weight/composition analysis.
Experimental groups
To investigate the role of α2AP on stasis thrombosis, we examined the following groups: α2AP+/+ mice; α2AP+/+ mice given 9 mg/kg of an anti-mouse inhibitory antibody sufficient to inhibit endogenous α2AP as reported24 ; α2AP−/− mice; α2AP−/− mice supplemented with a dose of human α2AP (4.2 mg/kg) sufficient to achieve physiological levels of α2AP as previously reported25 ; α2AP−/− mice given a physiological amount of α2AP (4.2 mg/kg) and equimolar amount of α2AP inhibiting antibody (9 mg/kg); α2AP−/− mice given a therapeutic dose of EACA (110 mg/kg); and Pg−/− mice. All treatments were given intravenously through jugular vein about 30 minutes before IVC ligation.
Tissue histology and staining
IVC tissue was fixed in 4% paraformaldehyde, paraffin-embedded and 5-µm cross sections were cut on slides. For staining, the IVC section slides were deparaffinized (Safeclear II, Fisher Diagnostics, MI) and hydrated by graded ethanol washes. Fibrin staining was done by Martius scarlet blue staining kit (Atom Scientific, Hyde, UK; #RRSK2-100) according to manufacturer instructions as described previously.26
For immunofluorescence staining of fibrinolytic system components (tPA, uPA, PAI-1, plasminogen, α2AP), the IVC tissue with or without thrombus was optimal cutting temperature-embedded after harvesting and 8-µm cryosections were cut on slides and air-dried. The sections on slides were fixed in chilled acetone for 15 minutes and again air-dried. The slides were washed with phosphate-buffered saline and blocked with 10% normal donkey serum for 1 hour. The sections were incubated with primary antibodies in 2% normal donkey serum for 90 minutes and then with secondary antibodies for 45 minutes. The primary antibodies include rabbit anti-mouse against tPA (ASMTPA-GF; Molecular Innovations, Novi, MI), uPA (ASMUPA-GF-HT; Molecular Innovations, Novi, MI), PAI-1 (IASMPAI-GF; Innovative Research, Novi, MI), plasminogen (Abcam; Cambridge, MA); goat anti-mouse α2AP (AF1239; R&D Systems, Minneapolis, MN); rat anti-mouse Ly6G (clone1A8, #127602; BioLegend, San Diego, CA), and Ly6B.2 (#771G; Bio-Rad Laboratories, Hercules, CA). Alexa Fluor 555 conjugated donkey anti-rabbit, donkey anti-goat, and Alexa Fluor 488 donkey anti-rat were used as secondary antibodies. The cell nuclei were stained with 4′,6-diamidino-2-phenylindole containing Vectashield hard-set mounting media (#H-1500; Vector Laboratories, Burlingame, CA). For blank controls, the primary antibody was not used on 1 section on each slide.
Imaging and analysis
All the experimental procedures including slide staining, scanning, imaging, and image analysis were done under the same conditions for all mice. Slides were digitally scanned by a blinded experimenter with Aperio image scanners, Aperio CS2 for brightfield, and Aperio FL for fluorescence imaging under same settings. The images were captured with freely available ImageScope software (Leica Biosystems) under constant settings. Whole IVC bright-field tissue images (original magnification ×4; 500 µm) were captured; representative magnified images (original magnification ×20; 200 µm) in the area of interest were selected under same brightness/contrast settings. For fluorescence imaging of tPA, uPA, plasminogen, PAI-1, and α2AP, the vascular wall of the IVC with or without thrombus in magnified (original magnification ×4) images (500 µm) was selected as an area of interest and quantified in Image-Pro Plus (Media Cybernetics, MD) in histogram mode with constant color range.
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis and immunoblotting of IVC thrombus
After 5 hours of IVC ligation in α2AP+/+ mice, the IVC containing the thrombus was harvested. The thrombus was separated from the vessel wall and solubilized in buffer (0.5 M Tris-HCl, pH 7.4, 1.5 M NaCl, 2.5% deoxycholic acid, 10% NP-40, and 10 mM EDTA with 1% sodium dodecyl sulfate). Protein was estimated by bicinchoninic acid assay using homogenized tissue supernatant after centrifugation. Proteins (100 µg) were electrophoresed on 10% reducing sodium dodecyl sulfate-polyacrylamide gel electrophoresis gels and transferred to a polyvinylidene difluoride membrane. The membrane was blocked with 10% bovine serum albumin in phosphate-buffered saline containing 0.1% Tween 20 and probed with rabbit anti-mouse plasminogen antibody followed by goat anti-rabbit 680 IRDye Li-COR secondary antibody (LI-COR Biosciences, NE). The blots were scanned by the LI-COR Odyssey infrared imaging system.
D-dimer assay
Because of the low sensitivity of available reagents for detecting mouse D-dimer, human fibrinogen (10 mg/kg in 200 µL saline) was given intravenously through the jugular vein to α2AP+/+ and α2AP−/− mice immediately before IVC ligation. A dose of 10 mg/kg fibrinogen to an adult mouse (with ∼2 mL blood) is expected to increase the fibrinogen concentration by ∼150 µg/mL (∼6% to 8% of endogenous fibrinogen, ∼2.5 mg/mL27 ), which is sufficient for the detection sensitivity of the kit (10 ng/mL). Five hours after IVC ligation, blood was collected by cardiac puncture to prepare plasma in 3.8% sodium citrate buffer (containing aprotinin). The vein thrombi weights in α2AP+/+ vs α2AP−/− mice were not significantly affected by this low-dose fibrinogen administration (∼6% of normal fibrinogen levels in mice) in comparison with their untreated genotypes (P = .62 and P = .10; Student t test), consistent with previous studies.27 D-dimer levels in plasma were quantitatively determined by Asserachrom D-Di Kit (#00947; Diagnostica Stago, Parsippany, NJ) according to the manufacturer’s instructions.
Statistical analysis
Statistical analysis was done with Graph Pad Prism 7.02 software (San Diego, CA). Data represent the mean ± standard error. The normality of data was tested by the Shapiro-Wilk test. Differences between 2 groups were compared by unpaired Student t test (for normally distributed data) or by Mann-Whitney U test. The differences among groups were analyzed by a one-way analysis of variance (ANOVA) analysis using the Newman-Keuls post hoc test. A two-tailed P < .05 was considered statistically significant.
Results
Expression of fibrinolytic system components in formed venous thrombi
To examine the role of fibrinolysis in thrombosis, we first determined whether the key components of the fibrinolytic system are present at the thrombus site. IVC sections of 24 hours (acute) and 7 days old (chronic) thrombi were immunostained (Figure 1) and compared with immunostained sham control veins (without IVC ligation). There was minimal tPA immunostaining in 24 hours and 7 days venous thrombi (Figure 1A-B). uPA was more prominent and present mostly within the thrombus (Figure 1C-D). Expression was mainly associated with neutrophils, which were present throughout the thrombus at 24 hours (supplemental Figure 1A-C, available on the Blood Web site). At 7 days, neutrophils largely colocalized with uPA expression (supplemental Figure 1D-F). Monocytes/macrophages (CD68+) were also present in the thrombus and vascular wall, and partially colocalized with uPA expression at 24 hours (supplemental Figure 2A-C). Macrophages were abundant in IVC thrombi at 7 days but showed limited colocalization with uPA expression (supplemental Figure 2D-F). PAI-1 was expressed in the vascular wall as well as at the thrombus-vein wall interface in 24-hour thrombi (Figure 1E) and it further increased over 7 days to included staining within the thrombus (Figure 1F). PAI-1 expression partially colocalized with platelets, which were present throughout the thrombus 24 hours (supplemental Figures 3A-C and 5D,F) and 7 days post-IVC ligation (supplemental Figures 3D-F and 5E,G). Plasminogen and α2AP immunostaining was 10- to 15-fold higher in the thrombus at 24 hours (Figure 1G-J) and 7 days by comparison with sham controls (Figure 1K-L; supplemental Figure 4). Fibrin(ogen) was also present throughout the thrombus at 24 hours and 7 days post-IVC ligation (supplemental Figure 5A-B,F,G).
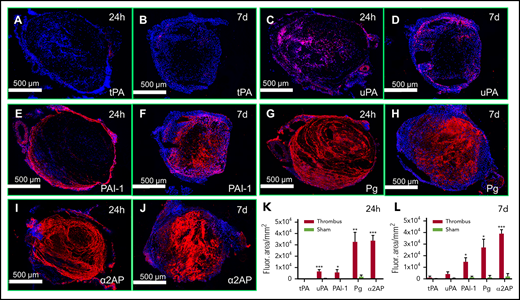
Expression of fibrinolytic system components in 24 h and 7 days old venous thrombi. Panels A-F show the expression (red) of (A-B) tPA, (C-D) uPA, (E-F) PAI-1, (G-H) plasminogen (Pg), (I-J) α2AP, and (K-L) their quantification in 8-µm cryosections of 24 hours and 7 days thrombi as labeled. Red color (Alexa Fluor 555) represents each component as labeled and blue color shows 4′,6-diamidino-2-phenylindole–stained nuclei. For comparison, IVC from sham mice were immunostained and quantified for each antibody under the same color range in the histogram mode of Image Pro-Plus software. The total immunostained area (arbitrary units; AU/mm2) for each protein was measured in 500 μm (original magnification ×4) images of an IVC sample. (N = 4-5 per group, mean ± SEM). *P < .05, **P < .01, ***P < .001.
Expression of fibrinolytic system components in 24 h and 7 days old venous thrombi. Panels A-F show the expression (red) of (A-B) tPA, (C-D) uPA, (E-F) PAI-1, (G-H) plasminogen (Pg), (I-J) α2AP, and (K-L) their quantification in 8-µm cryosections of 24 hours and 7 days thrombi as labeled. Red color (Alexa Fluor 555) represents each component as labeled and blue color shows 4′,6-diamidino-2-phenylindole–stained nuclei. For comparison, IVC from sham mice were immunostained and quantified for each antibody under the same color range in the histogram mode of Image Pro-Plus software. The total immunostained area (arbitrary units; AU/mm2) for each protein was measured in 500 μm (original magnification ×4) images of an IVC sample. (N = 4-5 per group, mean ± SEM). *P < .05, **P < .01, ***P < .001.
Early stasis thrombus formation
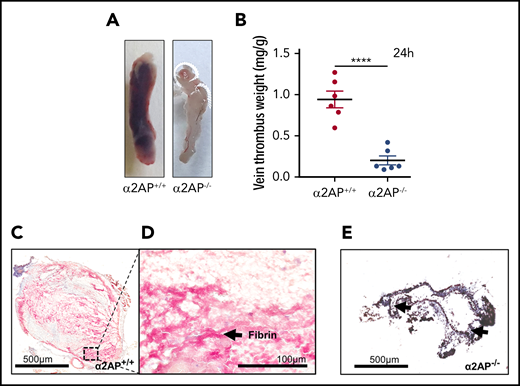
Thrombus growth and composition were examined after achieving full stasis by ligation of the main IVC and its side and back branches. Previous studies have shown that thrombi start developing as early as 3 to 5 hours of IVC ligation.21,28 Five hours after ligation, veins from wild-type mice were swollen and distended by red thrombus, in contrast to veins from sham mice who were subjected to surgery without ligation (Figure 2A). Consistent with their visual appearance, the combined vein thrombus weight in mice undergoing ligation was significantly increased compared with sham-operated mice (P < .0001) (Figure 2B). Martius scarlet blue staining of the veins showed a fibrin-rich thrombus containing red blood cells and leukocytes. The presence of fibrin(ogen) and neutrophils was also confirmed by immunofluorescence staining (supplemental Figure 6) as previously described21 (Figure 2C-D). Thrombi did not show any specific pattern of fibrin or cellular deposition, which is typical of early thrombosis.21
![Effects of α2AP deficiency on early thrombus formation. (A) Representative IVCs in different groups as labeled. (B) Vein thrombus weight milligram/gram of body weight. Each symbol represents an animal used in each group (N = 33). The bar graph shows the statistical significance between different groups by one-way ANOVA and Newman-Keuls post hoc test. ****P < .0001; ***P < .001; ns, nonsignificant. (C-F) Formalin-fixed paraffin-embedded (5 µm) sections of IVC thrombi were stained with Martius scarlet blue kit (scale bar = 500 µm). Representative image of N = 5 for each group. (C) Thrombus formation in the IVC of α2AP+/+ mice 5 hours after ligation (500 µm). (D) The composition of 5-hour-old thrombi (fibrin, red blood cell [RBC], and leukocytes; black arrows, scale bar = 100 µm). The pink/yellow color represents fibrin and nuclei are darkly stained. This panel shows a magnified image of the area in the black box highlighted in panel C. (E-F) Martius scarlet blue-stained representative images of IVC in α2AP−/− mice 5 hours after IVC ligation. The black arrows indicate areas of minimum thrombosis.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/134/12/10.1182_blood.2019000049/3/m_bloodbld2019000049f2.png?Expires=1769085834&Signature=eeAsNinbyOLvGmBCYPlahU-LlQCVSmvou7cvYhS28mCaQewNKbqropCZq-UCin-FmXMWp5yO-kumD1Dd5~Fs2xjHWu5lhXxv5NCmbhfTw3nhkz26A3wf8pyyoyhoZr-2EnBRmo0KpncaM6U01eynadYh9eseyDRiudTJfbznsQu-WXOHsXZcBocqxk5cpLMx6vq7DIiT~pihWmb6xR4mPgAj~HMYgci4u-iCO6mxTd1jM7M7C5SErgfoW-4pNU89pv3vAvWVpq1-IJQlby46ZciSQEOoKTki4DVSY5fUQ-Sr4t0I7tpxaKFdtsxqzeyg8x3leBqNtVBQal7odx0Mag__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Effects of α2AP deficiency on early thrombus formation. (A) Representative IVCs in different groups as labeled. (B) Vein thrombus weight milligram/gram of body weight. Each symbol represents an animal used in each group (N = 33). The bar graph shows the statistical significance between different groups by one-way ANOVA and Newman-Keuls post hoc test. ****P < .0001; ***P < .001; ns, nonsignificant. (C-F) Formalin-fixed paraffin-embedded (5 µm) sections of IVC thrombi were stained with Martius scarlet blue kit (scale bar = 500 µm). Representative image of N = 5 for each group. (C) Thrombus formation in the IVC of α2AP+/+ mice 5 hours after ligation (500 µm). (D) The composition of 5-hour-old thrombi (fibrin, red blood cell [RBC], and leukocytes; black arrows, scale bar = 100 µm). The pink/yellow color represents fibrin and nuclei are darkly stained. This panel shows a magnified image of the area in the black box highlighted in panel C. (E-F) Martius scarlet blue-stained representative images of IVC in α2AP−/− mice 5 hours after IVC ligation. The black arrows indicate areas of minimum thrombosis.
Effects of α2AP deficiency on early thrombus formation. (A) Representative IVCs in different groups as labeled. (B) Vein thrombus weight milligram/gram of body weight. Each symbol represents an animal used in each group (N = 33). The bar graph shows the statistical significance between different groups by one-way ANOVA and Newman-Keuls post hoc test. ****P < .0001; ***P < .001; ns, nonsignificant. (C-F) Formalin-fixed paraffin-embedded (5 µm) sections of IVC thrombi were stained with Martius scarlet blue kit (scale bar = 500 µm). Representative image of N = 5 for each group. (C) Thrombus formation in the IVC of α2AP+/+ mice 5 hours after ligation (500 µm). (D) The composition of 5-hour-old thrombi (fibrin, red blood cell [RBC], and leukocytes; black arrows, scale bar = 100 µm). The pink/yellow color represents fibrin and nuclei are darkly stained. This panel shows a magnified image of the area in the black box highlighted in panel C. (E-F) Martius scarlet blue-stained representative images of IVC in α2AP−/− mice 5 hours after IVC ligation. The black arrows indicate areas of minimum thrombosis.
To determine the potential role of fibrinolysis in stasis-induced thrombosis, we examined congenic mice that were deficient in α2AP. Vein thrombus weight was significantly less in α2AP−/− mice than in wild-type (α2AP+/+) mice (P < .0001); there was no significant difference between vein thrombus weights from α2AP−/− mice and sham mice (Figure 2A-B). Martius scarlet blue staining of the veins from α2AP−/− mice showed minimal thrombus formation (Figure 2E-F). Because thrombus development was severely hampered in α2AP−/− mice, a minimum number of red blood cells in the vessels of α2AP−/− mice were observed. Red blood cells were present throughout the thrombus in α2AP+/+ mice (supplemental Figure 7). The vein thrombus weight in α2AP+/+ mice was significantly reduced by treatment with an α2AP inactivating (α2AP-I) antibody24,25 (9 mg/kg) compared with untreated α2AP+/+ mice (P < .001); the effects of the α2AP-I were equivalent to those seen in α2AP−/− or sham mice (Figure 2B). There were no significant sex-related differences in vein thrombus weight in normal wild-type (α2AP+/+) male vs female mice (P = .57, N = 4-7), 5 hours after IVC ligation (data not shown).
Vein thrombus weight 24 hours following ligation was significantly greater than 5 hours after ligation (P = .004). Thrombi formed 24 hours after ligation extended throughout the entire length of the IVC in α2AP+/+ mice (Figure 3A). Martius scarlet blue staining suggested that thrombi emanated from the vascular wall with fibrin evident throughout the thrombus (Figure 3C-D). The vein thrombus weight was significantly reduced in α2AP−/− mice in comparison with α2AP+/+ mice (P < .0001) (Figure 3A-B,E). Small areas of thrombosis were observed in veins from α2AP−/− mice by Martius scarlet blue staining (Figure 3E). Microscopically, a thin layer of platelets (supplemental Figure 8B) was observed on the vascular endothelium in α2AP−/− mice by immunofluorescence staining (supplemental Figure 8A-C), which was less than the extensive platelet deposition seen in α2AP+/+ mice (supplemental Figure 5D,F). Immunostaining of the microscopic platelet thrombi in α2AP−/− mice also revealed the presence of fibrin(ogen) (supplemental Figure 8D-F). To further examine if partial blood flow affects the contribution of α2AP to deep vein thrombosis, IVC stenosis was induced in α2AP+/+ mice vs α2AP−/− mice for 24 hours. As expected for the IVC stenosis model, there was greater variation in vein thrombus weight. However, the median vein thrombus weight was significantly reduced in α2AP−/− mice vs α2AP+/+ mice (P < .01) (supplemental Figure 8G-H).
Effects of α2AP deficiency on acute thrombus formation. (A-B) Representative IVCs in α2AP+/+ and α2AP−/− mice (A) 24 hours after IVC ligation and (B) their thrombus weights. Each symbol in the bar graph represents an animal used in each group (N = 12); ****P < .0001, Student t test. (C) Martius scarlet blue (scale bar = 500 µm) staining shows 24-hour thrombi rich in fibrin (pink color). (D) Magnified image (original magnification ×20; scale bar = 100 µm) of panel C showing a fibrin-rich (pink) thrombus area. (E) Minimum thrombus formation (black arrows) in α2AP−/− mice 24 hours after IVC ligation.
Effects of α2AP deficiency on acute thrombus formation. (A-B) Representative IVCs in α2AP+/+ and α2AP−/− mice (A) 24 hours after IVC ligation and (B) their thrombus weights. Each symbol in the bar graph represents an animal used in each group (N = 12); ****P < .0001, Student t test. (C) Martius scarlet blue (scale bar = 500 µm) staining shows 24-hour thrombi rich in fibrin (pink color). (D) Magnified image (original magnification ×20; scale bar = 100 µm) of panel C showing a fibrin-rich (pink) thrombus area. (E) Minimum thrombus formation (black arrows) in α2AP−/− mice 24 hours after IVC ligation.
Thrombus formation during prolonged venous stasis
When compared with the early thrombus, ligation of the IVC in α2AP+/+ mice for 7 days produced a lamellated thrombus, containing successive layers of fibrin typical of mature thrombi14,21 (Figure 4A-B). Fourteen days following IVC ligation in α2AP+/+ mice, there was still evidence of obstructive fibrin thrombi at the center of the IVC, but the pattern appeared more “moth-eaten” or porous (Figure 4D-E). In contrast, minimum microscopic thrombus formation was visible in α2AP−/− mice at 7 days or 14 days after ligation (Figure 4C,F). In α2AP+/+ mice, there was a significant increase in vein thrombus weight from 5 hours to 7 days (twofold, P < .0001) that decreased from 7 to 14 days (P < .01; Figure 4H; supplemental Figure 8I). Vein thrombus weight was greater in α2AP+/+ mice than sham mice at all the time points (P < .0001; Figure 4G-H). Vein thrombus weights were markedly reduced in α2AP−/− vs α2AP+/+ mice at 7 and 14 days (P < .0001; Figure 4G-H). There was no significant difference between vein weights from sham and α2AP−/− mice after prolonged ligation (Figure 4H), indicating that α2AP deficiency was associated with prolonged suppression of stasis-induced venous thrombosis at 7 and 14 days, just as it prevented early thrombosis (supplemental Figure 8I).
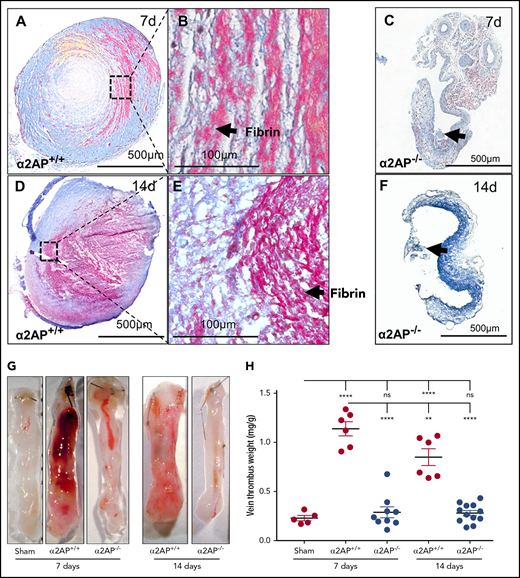
Effects of α2AP deficiency on chronic thrombus formation. (A) Martius scarlet blue-stained 7-day-old thrombi (scale bar = 500 µm). (B) Magnified images (original magnification ×20) of IVC thrombus showing lamellate fibrin-rich (pink) lines of Zahn. (C) Thrombi were not formed in α2AP−/− mice after 7 days of IVC ligation. Arrow indicates the minimum thrombus formation. (D) Martius scarlet blue stained 14 days old thrombi (scale bar = 500 µm) in α2AP+/+ mice. (E) Magnified image (original magnification ×20) of panel D showing a fibrin-rich (pink), porous thrombus. (F) Martius scarlet blue-stained IVC showing minimum thrombus in α2AP−/− mice. (G) Representative images of IVC thrombus formation in different groups as labeled including sham mice (7 days). (H) Vein thrombus weight milligram/gram of body weight. Each symbol represents an animal used in each group (N = 38). One-way ANOVA; ****P < .0001, **P < .01.
Effects of α2AP deficiency on chronic thrombus formation. (A) Martius scarlet blue-stained 7-day-old thrombi (scale bar = 500 µm). (B) Magnified images (original magnification ×20) of IVC thrombus showing lamellate fibrin-rich (pink) lines of Zahn. (C) Thrombi were not formed in α2AP−/− mice after 7 days of IVC ligation. Arrow indicates the minimum thrombus formation. (D) Martius scarlet blue stained 14 days old thrombi (scale bar = 500 µm) in α2AP+/+ mice. (E) Magnified image (original magnification ×20) of panel D showing a fibrin-rich (pink), porous thrombus. (F) Martius scarlet blue-stained IVC showing minimum thrombus in α2AP−/− mice. (G) Representative images of IVC thrombus formation in different groups as labeled including sham mice (7 days). (H) Vein thrombus weight milligram/gram of body weight. Each symbol represents an animal used in each group (N = 38). One-way ANOVA; ****P < .0001, **P < .01.
Stasis-induced venous thrombosis requires α2AP
The finding that α2AP deficiency blocked the formation of venous thrombosis suggested that α2AP itself or compensatory physiologic changes induced by α2AP deficiency were essential for thrombus development. Infusion of human α2AP to α2AP−/− mice in doses sufficient to achieve physiologic levels of α2AP (mean, 58 ± 8 µg/mL),25 restored stasis-induced thrombus formation 5 hours after ligation vs α2AP−/− mice (P < .0001) to levels comparable to that observed in α2AP+/+ mice (Figure 5A-B). However, when human α2AP was given to α2AP−/− mice, followed by infusion of a monoclonal antibody α2AP-I, which inactivates human α2AP, thrombus formation was significantly reduced vs α2AP−/− mice supplemented with α2AP to levels observed in α2AP−/− mice (Figure 5A-B).24,25,29 These complementary data demonstrate that stasis-induced venous thrombosis specifically requires α2AP.
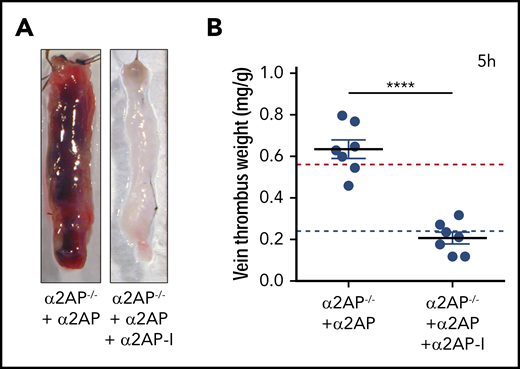
Effects of α2AP supplementation/inhibition. (A) Representative images of IVC thrombus formation in α2AP−/− mice after 5 hours’ ligation after administration of α2AP or α2AP + α2AP-inactivating antibody (α2AP-I) as labeled. (B) Vein thrombus weight in these groups in milligram/gram of body weight as labeled. Each symbol represents a single experiment, N = 14. The horizontal red and blue lines in the graph show, for reference, the mean thrombus weight in α2AP+/+ and α2AP−/− mice, respectively. ****P < .0001; one-way ANOVA (among 4 groups).
Effects of α2AP supplementation/inhibition. (A) Representative images of IVC thrombus formation in α2AP−/− mice after 5 hours’ ligation after administration of α2AP or α2AP + α2AP-inactivating antibody (α2AP-I) as labeled. (B) Vein thrombus weight in these groups in milligram/gram of body weight as labeled. Each symbol represents a single experiment, N = 14. The horizontal red and blue lines in the graph show, for reference, the mean thrombus weight in α2AP+/+ and α2AP−/− mice, respectively. ****P < .0001; one-way ANOVA (among 4 groups).
Effects of α2AP on venous thrombus formation are mediated through fibrinolysis
α2AP is the primary inhibitor of the fibrinolytic enzyme plasmin, which cleaves cross-linked fibrin to form the soluble fibrin product D-dimer. Early venous stasis thrombi contained plasminogen and plasminogen activators (Figure 1), with evidence of plasmin generation, as detected by immunoblotting of thrombus lysate (supplemental Figure 9). If stasis initiates fibrin formation, which triggers plasmin-mediated fibrinolysis to block thrombus formation, IVC ligation should increase D-dimer levels in mice lacking α2AP. To examine this, D-dimer levels in plasma were determined after IVC ligation in α2AP+/+ and α2AP−/− mice as described in Methods. Five hours after venous ligation, α2AP−/− mice showed a nearly fivefold increase in D-dimer levels by comparison with α2AP+/+ mice (P < .05; Figure 6A), signifying enhanced fibrinolysis. As expected, vein thrombus weights were significantly reduced in α2AP−/− vs α2AP+/+ mice (P < .001; Figure 6B).
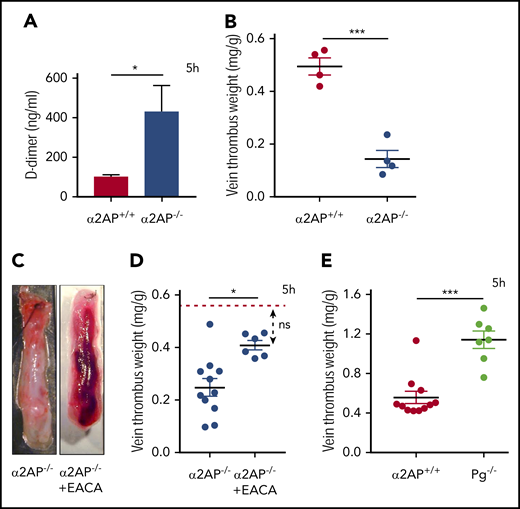
Effects of α2AP on fibrinolysis. (A) Plasma D-dimer levels in α2AP+/+ and α2AP−/− mice after 5 hours of IVC ligation. The mice were supplemented with 10 mg/kg human fibrinogen. *P < .05 (Student t test). (B) Vein thrombus weight milligram/gram of body weight in α2AP+/+ and α2AP−/− mice after 5 hours’ IVC ligation used for D-dimer measurements ***P < .001 (Student t test). (C) Representative images of IVC thrombus formation in EACA-treated α2AP−/− mice 5 hours after ligation as labeled. (D) Vein thrombus weight (milligram/gram of body weight) in α2AP−/− mice with or without EACA treatment. The horizontal red line in the graph shows the mean vein thrombus weight in α2AP+/+ mice for reference. *P < .05, one-way ANOVA (among 3 groups). (E) Vein thrombus weight (milligram/gram of body weight) in Pg−/− mice in comparison with α2AP+/+ mice. (N = 21) ***P < .001, Mann-Whitney test.
Effects of α2AP on fibrinolysis. (A) Plasma D-dimer levels in α2AP+/+ and α2AP−/− mice after 5 hours of IVC ligation. The mice were supplemented with 10 mg/kg human fibrinogen. *P < .05 (Student t test). (B) Vein thrombus weight milligram/gram of body weight in α2AP+/+ and α2AP−/− mice after 5 hours’ IVC ligation used for D-dimer measurements ***P < .001 (Student t test). (C) Representative images of IVC thrombus formation in EACA-treated α2AP−/− mice 5 hours after ligation as labeled. (D) Vein thrombus weight (milligram/gram of body weight) in α2AP−/− mice with or without EACA treatment. The horizontal red line in the graph shows the mean vein thrombus weight in α2AP+/+ mice for reference. *P < .05, one-way ANOVA (among 3 groups). (E) Vein thrombus weight (milligram/gram of body weight) in Pg−/− mice in comparison with α2AP+/+ mice. (N = 21) ***P < .001, Mann-Whitney test.
To further determine if the effects of α2AP deficiency were mediated through fibrinolysis, α2AP−/− mice were treated with a clinical dose of EACA (110 mg/kg), which blocks the fibrinolytic action of plasmin.30,31 EACA significantly reversed the effects of α2AP deficiency on thrombus formation (Figure 6C-D). Thrombus vein weights in EACA-treated α2AP−/− mice were significantly enhanced by comparison with untreated α2AP−/− mice (P < .05) and were not significantly different from α2AP+/+ mice (Figure 6C-D). To confirm that stasis induces plasmin-mediated fibrinolysis to affect venous thrombosis, we examined thrombus weights in mice lacking fibrinolysis because of deficiency of plasminogen (Pg−/−). Five hours after IVC ligation, vein thrombus weights in Pg−/− mice were significantly larger than those in congenic α2AP+/+ mice (P < .001, Figure 6E). These data suggest that stasis induces plasmin-mediated fibrinolysis that must be suppressed by α2AP to enable the development of vein thrombosis.
Discussion
Experimental venous stasis induces the formation of the large, stable venous thrombi, which grow over the course of a week and persist for at least 14 days. The early initiation of stasis thrombosis, as well as its acute and chronic development, requires α2AP because significant thrombus formation was blocked at 5 hours, 24 hours, 7 days, and 14 days after IVC ligation in α2AP−/− mice. The finding that D-dimer levels rise acutely in α2AP−/− mice after IVC ligation suggests that venous stasis activates the fibrinolytic system to prevent thrombus formation. Consistent with this, thrombus formation could be restored in α2AP−/− mice by blocking fibrinolysis with the indirect plasmin inhibitor EACA or by administering α2AP. Moreover, thrombus formation was significantly greater in mice with suppressed fibrinolysis resulting from plasminogen deficiency. Besides complete IVC stasis model, α2AP deficiency also suppresses the thrombus formation in the IVC stenosis model that mimics partial blood flow conditions sometimes observed in clinical settings. Taken together, these data suggest that venous stasis induces thrombosis and activates fibrinolysis, which must be suppressed by α2AP, to enable venous thrombus formation. It is interesting to note that although Virchow noted stasis and coagulation as important factors in thrombosis, the role of fibrinolysis suppression in this process has not received much recognition.
In vitro measures of diminished endogenous fibrinolysis (hypofibrinolysis) have been associated with increased risk of human venous thrombosis32 in blood samples obtained from patients a median of 10 months postevent.19 In these in vitro tests, reduced fibrinolytic potential has been mainly attributed to increased PAI-1 and thrombin-activable fibrinolysis inhibitor (TAFI). Surprisingly, neither increased levels of α2AP nor plasminogen deficiency has been identified as strong risk factors for future venous thrombosis in human plasma clot lysis assays. Stasis-induced thrombosis in vivo is considered highly resistant to the effects of fibrinolysis and as such, it is a rigorous test of the potential role of fibrinolysis in thrombus development.20 In deep vein thrombosis induced by IVC stenosis, tPA−/− and uPA−/− mice did not show any differences from control mice in the development of early thrombosis.16 However, thrombus dissolution after IVC ligation is reduced in uPA−/− mice, but not in tPA−/− mice, in comparison with wild-type controls.16 uPA was present throughout the thrombus, colocalized with neutrophils (and to a less extent, monocytes/macrophages), suggesting it may play a major role in driving fibrinolysis. There was evidence for plasmin generation and plasmin-triggered fibrinolysis in the early phase of stasis-induced thrombosis and plasminogen levels affected the extent of early stasis-induced thrombosis. However, plasmin generation during early thrombosis appeared insufficient to overcome the antifibrinolytic effects of α2AP. Deficiency of α2AP promoted fibrinolysis and reduced both early (5 hours) and later (up to 14 days) thrombus formation. Early inhibition of α2AP activity also significantly reduced thrombus formation; the effect of longer term α2AP inhibition on chronic thrombosis was not determined. PAI-1 deficiency moderately reduced thrombosis 2 or 8 days (∼25%) after IVC stasis.15,33 PAI-1 has also been shown to accentuate endotoxin-initiated hindfoot thrombosis in mice.34,35 Although these reports do not specifically address the early initiation of fibrinolysis during venous stasis, they provide confirmation that the fibrinolytic process modifies the extent or eventual volume of venous thrombosis. Still, current studies do not provide a full understanding of how fibrinolytic inhibitors such as α2AP, PAI-1, PAI-2, and TAFI may work together to regulate thrombus formation during venous stasis. Further research is also needed to determine how other critical variables such as age, sex, and partial venous occlusion may modify thrombus formation and fibrinolysis.
Enhancing fibrinolysis to prevent or treat thrombosis may raise concerns about potential hemorrhagic complications. Although α2AP−/− mice have enhanced fibrinolytic potential, they do not show excessive bleeding (experimental tail hemorrhage) with36 or without a thrombotic challenge.37 Recent studies show that complete deficiency of α2AP and its therapeutic inactivation enhances thrombus dissolution without increasing bleeding risk in experimental ischemic stroke24 and pulmonary embolism, even in the presence of plasminogen activators.25 Fibrinogen, plasminogen, PAI-1, or other hemostasis parameters remain unchanged in α2AP−/− mice.37 Early-phase human clinical trials also show that α2AP inhibition increases D-dimer levels in healthy volunteers without adverse events.38
Taken together, these data suggest venous stasis induced by ligation activates the fibrinolytic system. Suppression of plasmin-induced fibrinolysis by α2AP plays an essential role in enabling the formation of venous thrombosis for a period of up 14 days, the latest time point in our studies. These data suggest that targeting α2AP may prove useful for preventing the initial development of venous thrombosis as well as for resolving already formed thrombi.
For original data, please contact guyreed@email.arizona.edu.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors appreciate the technical contributions of Nelson Houng.
This work was funded in part by grants from the National Institutes of Health, National Institute of Neurological Disorders and Stroke (NS089707) and the National Institutes of Health, National Heart, Lung, and Blood Institute (HL092750) to G.L.R.
Authorship
Contribution: S.S. and A.K.H. performed the experiments, analyzed data, and wrote the manuscript; and G.L.R. designed the research, analyzed the data, and wrote the manuscript.
Conflict-of-interest disclosure: G.L.R is a founder of Translational Sciences. The remaining authors declare no competing financial interests.
Correspondence: Guy L. Reed, 475 N. 5th St, The University of Arizona, College of Medicine, Phoenix, AZ 85004; e-mail: guyreed@email.arizona.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal