

Key Points
The safety profile of ibrutinib, lenalidomide, and rituximab in combination was manageable in patients with relapsed/refractory DLBCL.
The combination demonstrated promising and durable activity in relapsed/refractory DLBCL, particularly in patients with non-GCB DLBCL.

Abstract
The outcome of patients with relapsed/refractory diffuse large B-cell lymphoma (DLBCL) is poor, particularly in patients ineligible for stem cell transplantation or who fail induction therapy or salvage therapy. The phase 1b portion of this open-label, dose-escalation (3+3+3 design) study examined the maximum tolerated dose (MTD) and preliminary safety and activity of the regimen in transplant-ineligible adults with histologically confirmed relapsed/refractory DLBCL after at least 1 prior therapy. Patients received once-daily 560 mg ibrutinib, 375 mg/m2 intravenous rituximab day 1 of cycles 1 to 6, and 10, 15, 20, or 25 mg lenalidomide days 1 to 21 of each 28-day cycle. Forty-five patients were treated; median time since diagnosis was 14.1 months, and 51% of the patients had non–germinal center B-cell–like (non-GCB) DLBCL, 33% had transformed DLBCL, 60% were refractory, and 27% were primary refractory. Because of dose-limiting toxicities, a de-escalation cohort (10 mg lenalidomide) was initiated, and with subsequent re-escalation up to 25 mg lenalidomide, the MTD was not reached. In response-evaluable patients, the overall response rate (ORR) was 44% (complete response [CR], 28%); among them, the ORR was 65% (CR, 41%) in non-GCB and 69% and 56% in relapsed (n = 16) and secondary refractory (n = 27) disease, respectively. Overall and for non-GCB, median response duration was 15.9 months, with 2 patients receiving therapy beyond 3 years. Phase 2 was initiated with 20 mg lenalidomide in relapsed/refractory non-GCB, whereas the phase 1b 25-mg lenalidomide cohort was being completed; an additional 25-mg cohort in phase 2 is currently ongoing. This study was registered at www.clinicaltrials.gov as #NCT02077166.
Introduction
Diffuse large B-cell lymphoma (DLBCL) is the most common form of non-Hodgkin lymphoma and is increasing in incidence, particularly for elderly patients.1,-3 The R-CHOP regimen (rituximab with cyclophosphamide, doxorubicin, vincristine, and prednisone) is curative for more than half of patients with DLBCL.1,2,4 Although the standard of care in patients who fail first-line chemoimmunotherapy is salvage therapy followed by high-dose therapy and stem cell transplantation, only 10% to 20% of patients have long-term disease-free survival.1,2,4,5 Patients with primary refractory disease or who relapse within 12 months have a dismal outcome with the current standard of care; median overall survival (OS) is about 5 to 6 months.1,2,4 Moreover, patients may be ineligible for transplantation because of their age or comorbidities. Patients who relapse after autologous stem cell transplantation also have poor outcomes with limited treatment options.1,2,4 Chimeric antigen receptor T-cell therapy shows promise in these patients; however, this therapy is limited to certain treatment centers and has potentially limiting toxicity leading to intolerability for some patients.6 Therefore, there is a substantial unmet need for novel treatments for relapsed/refractory DLBCL.
Ibrutinib, a first-in-class, once-daily inhibitor of Bruton’s tyrosine kinase, is indicated by the US Food and Drug Administration for a variety of B-cell malignancies and graft-versus-host disease.7 Inhibition of Bruton’s tyrosine kinase disrupts chronic B-cell receptor (BCR) signaling, leading to decreased nuclear factor-κB activity.8 Lenalidomide is a once-daily thalidomide analog indicated by the US Food and Drug Administration for multiple myeloma in combination with dexamethasone, for mantle cell lymphoma after relapse or progression after 2 prior therapies including bortezomib, and for transfusion-dependent anemia resulting from myelodysplastic syndrome.9 Lenalidomide is an immunomodulatory agent that down-modulates interferon regulatory factor 4 downstream of BCR and myeloid differentiation primary response 88 (MYD88) signaling, leading to increased interferon-β secretion and decreased nuclear factor-κB activity. Preclinical studies suggest increased activity in DLBCL models when ibrutinib and lenalidomide are combined, particularly in non–germinal center B-cell–like (non-GCB) and activated B-cell (ABC) DLBCL, which depend on both the BCR and MYD88 signaling pathways, downstream activation of nuclear factor-κB, and upregulation of interferon regulatory factor 4 expression for survival.8 Both drugs have demonstrated activity in relapsed/refractory non-GCB and ABC DLBCL.10,,,,,-16 Rituximab, a chimeric monoclonal antibody that binds CD20 on B cells, is approved by the US Food and Drug Administration for non-Hodgkin lymphoma and chronic lymphocytic leukemia17 ; based on its B-cell target, rituximab may provide additional activity to ibrutinib plus lenalidomide, with data suggesting lenalidomide’s immunomodulatory properties (increased natural killer cells and T-cell activation and improved immunological synapses)18 may overcome rituximab resistance in B-cell lymphomas.19 The combination of ibrutinib and rituximab has also shown promising activity, particularly in mantle cell lymphoma.20
The combination of ibrutinib, lenalidomide, and rituximab was recently studied in a phase 1 study in adults with previously untreated follicular lymphoma (FL).21 Adding ibrutinib did not provide improved activity over prior reports of lenalidomide and rituximab and revealed a more severe toxicity profile than expected (particularly rash: all grades in 82% of cases and grade 3 in 36% of cases). Although no dose-limiting toxicities (DLTs) were observed, the authors concluded that the observed toxicity of the regimen did not warrant further investigation in patients with FL. However, FL is a relatively slow-growing lymphoma, and in the front-line setting, many treatment options are available. Conversely, DLBCL is a rapidly growing lymphoma with few viable treatment options in the relapsed/refractory setting, particularly among the elderly. Therefore, the current study evaluated the combination of ibrutinib, lenalidomide, and rituximab in transplant-ineligible adults with relapsed/refractory DLBCL.
Patients and methods
Study design
Study PCYC-1123-CA (registered at www.clinicaltrials.gov as NCT02077166) is an open-label, multicenter, phase 1b/2 study. The phase 1b dose-escalation portion, presented here, aimed to establish the maximum tolerated dose and evaluate the safety and tolerability of ibrutinib plus lenalidomide and rituximab in transplant-ineligible patients with relapsed/refractory DLBCL.
Patients received fixed doses of oral 560 mg ibrutinib daily throughout each 28-day treatment cycle (dose established based on data from a prior phase 1/2 study),16 375 mg/m2 intravenous (IV) rituximab on day 1 of cycles 1 to 6, and escalating doses of lenalidomide on days 1 to 21 of each cycle (Figure 1A). Patients completed an end-of-treatment visit 30 days after treatment discontinuation and then were monitored for OS and subsequent anticancer therapies. Patients who discontinued for reasons other than progressive disease (PD) or death also had disease evaluations every 12 weeks (±7 days) until PD or start of subsequent anticancer therapies.
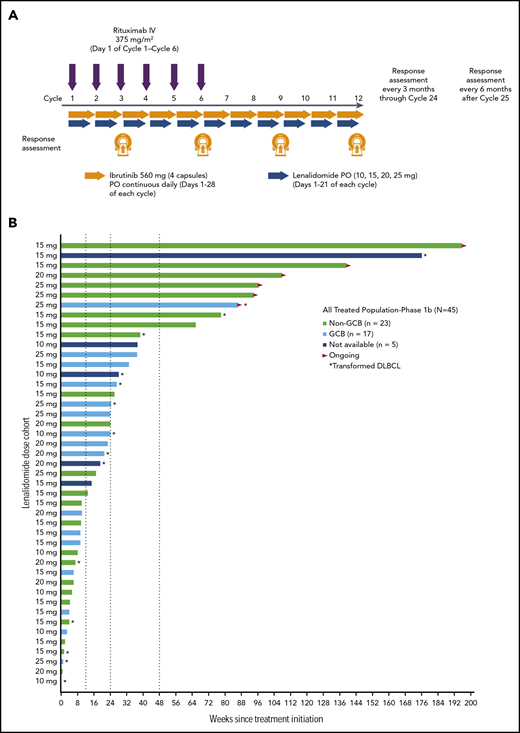
Phase 1b schema and treatment duration. The phase 1b treatment schema is presented in panel A. Data on treatment duration are presented in panel B for patients in the all-treated population, with each bar representing an individual patient. Patients are ordered by the study initial dose date and color-coded by DLBCL subtype. The indicated lenalidomide dose was given in combination with 560 mg/day ibrutinib and 375 mg/m2 IV rituximab. PO, per os.
Phase 1b schema and treatment duration. The phase 1b treatment schema is presented in panel A. Data on treatment duration are presented in panel B for patients in the all-treated population, with each bar representing an individual patient. Patients are ordered by the study initial dose date and color-coded by DLBCL subtype. The indicated lenalidomide dose was given in combination with 560 mg/day ibrutinib and 375 mg/m2 IV rituximab. PO, per os.
Phase 1b dose escalation followed traditional 3+3+3 principles22 ; maximum tolerated dose was defined as the highest dose at which 33% or less of patients who were enrolled in a cohort experienced a DLT. Hematologic DLTs were defined as the following events that are considered possibly related to study drug: grade 4 neutropenia (absolute neutrophil count <500/mm3) lasting more than 7 days, life-threatening grade 3 or higher neutropenia (absolute neutrophil count <1000/mm3) with fever at least 38.3°C, grade 4 thrombocytopenia (platelets <25 000/mm3) lasting at least 7 days despite holding treatment, and grade 3 thrombocytopenia with grade 2 or higher bleeding or requiring red blood cell or platelet transfusion. Nonhematologic DLTs were defined as any grade 3 or higher adverse event (AE) except grade 3 tumor lysis syndrome, unless it required dialysis; grade 3 deep venous thrombosis, unless it was unresponsive to anticoagulation; grade 3 rash, unless it has not improved within 10 days to grade 1; grade 3 infection; grade 3 or higher nausea, vomiting, or diarrhea, unless uncontrolled by maximal supportive care and persisting more than 7 days; and grade 3 fatigue lasting more than 7 days.
The sponsor and investigators developed management strategies for specific AEs during the study conduct. For grade 3 or higher rash, ibrutinib was withheld until improvement to grade 1 and then resumed at the original or a lower dose; treatment with 20 to 100 mg of oral prednisone (or equivalent) daily for approximately 1 to 5 weeks per investigator’s discretion (with or without taper) and oral antihistamines were recommended. For grade 3 neutropenia with a temperature of at least 38.5°C and grade 4 neutropenia lasting more than 7 days, ibrutinib was withheld until recovery to an absolute neutrophil count of at least 750/mm3 and resumed at the original (first occurrence) or next lower dose (subsequent occurrences), and lenalidomide was withheld for the remainder of the treatment cycle and resumed at the next lower dose; hematopoietic growth factors were allowed. Ibrutinib and lenalidomide could be withheld for 28 or fewer consecutive days for toxicity.
Study population
Eligible patients were at least 18 years of age with relapsed/refractory pathologically confirmed DLBCL with 1 or more measurable disease sites larger than 1.5 cm in the longest dimension on a computed tomography scan. Relapsed/refractory status was defined as recurrence of disease after complete response (CR) or partial response (PR), stable disease, or PD at completion of the preceding regimen (including autologous transplantation). Patients without prior transplant must have been transplant-ineligible at study entry, defined as 1 or more criterion: age at least 70 years, diffuse lung capacity for carbon monoxide lower than 50%, left ventricular ejection fraction lower than 50%, organ dysfunction or comorbidities precluding transplantation, failure to achieve CR or PR with salvage therapy, or patient refusal. Patients were required to have the ability to swallow capsules; an Eastern Cooperative Oncology Group performance status lower than 2; life expectancy higher than 3 months; adequate hematologic, hepatic, and renal function; and both a prothrombin time/international normalized ratio and activated partial thromboplastin time of less than 1.5 × the upper limit of normal.
Key exclusion criteria included unresolved toxicities from prior anticancer therapy; medically apparent central nervous system lymphoma/leptomeningeal disease; prior allogeneic transplantation, other malignancies (except adequately treated nonmelanoma skin cancer), or HIV or hepatitis B or C infection; recent infection requiring IV anti-infective treatment completed within 14 days of study drug; known bleeding diathesis or hemophilia; clinically significant cardiovascular disease; and known hypersensitivity to study drug. Patients must not have received any chemotherapy, external beam radiation therapy, or anticancer antibodies within 2 weeks of study drug; radio- or toxin-immunoconjugates within 10 weeks of study drug; systemic immunosuppressant therapy within 28 days of study drug; or major surgery within 4 weeks of study drug.
Assessments and end points
Safety was assessed through physical examinations, vital sign measurements, laboratory tests, and AE reporting. Treatment-emergent AEs (TEAEs) were reported from treatment initiation through 30 days after treatment discontinuation. The investigators attributed causality.
Non-GCB or GCB phenotype was determined by immunohistochemistry, using the Hans algorithm.23 In patients with sufficient tissue, gene expression profiling was performed to further classify patients as ABC, GCB, or unclassified. Computed tomography, magnetic resonance imaging, and/or positron emission tomography scans were performed within 28 days before the first dose (baseline), within 7 days before day 1 of cycles 4 and 7, and every 3 cycles thereafter, up to cycle 25. Efficacy end points included overall response rate (ORR) and CR rate. The investigators assessed tumor response, using the revised International Working Group response criteria for non-Hodgkin lymphoma.24
Study oversight
The study was conducted in compliance with Good Clinical Practice, as described in the International Conference on Harmonization and according to applicable regulatory requirements. The study protocol and all amendments were approved by applicable institutional review boards/independent ethics committees, and all patients provided written informed consent before enrollment. The sponsor analyzed study data; all authors had access to all primary clinical study data and participated in data interpretation.
Results
Patients
Forty-five patients were enrolled in the phase 1b portion and received ibrutinib and rituximab plus 10, 15, 20, or 25 mg lenalidomide. Patient demographics and baseline clinical characteristics were similar across dose cohorts, with some variations resulting from the small patient numbers (Table 1). The median age was 64 years, with a median time since diagnosis of 14.1 months. Approximately half (51%) the patients had non-GCB DLBCL and 33% had transformed DLBCL (7/15 with GCB DLBCL, 5/15 with non-GCB DLBCL, and 3/15 without classification). Of 15 patients with transformed DLBCL, 13/15 had prior FL, 1/15 had prior chronic lymphocytic leukemia, and 1/15 had prior marginal zone lymphoma. Three patients had DLBCL with rearrangements of MYC and BCL2 and/or BCL6. Most (84%) patients had received at least 2 prior regimens, and 60% of patients had refractory disease, including 27% of patients who had primary refractory DLBCL. For all 45 enrolled transplant-ineligible patients, 21 had failed to achieve a response with salvage therapy, 13 were at least 70 years of age, 9 had prior autologous transplant, and 2 refused transplant.
Baseline demographic and clinical characteristics
| 560 mg/day ibrutinib + 375 mg/m2 IV rituximab combined with: | |||||||
|---|---|---|---|---|---|---|---|
| Dose level −1: 10 mg LEN (n = 7) | Dose level 1: 15 mg LEN (n = 12) | Dose level 1+: 15 mg LEN (n = 9) | Dose level 2: 20 mg LEN (n = 9) | Dose level 3: 25 mg LEN (n = 8) | Dose level 1 combined (n = 21) | All dose levels (N = 45) | |
| Age | |||||||
| Median age, y (range) | 59 (49-68) | 62 (43-84) | 68 (41-85) | 64 (50-77) | 68 (36-78) | 64 (41-85) | 64 (36-85) |
| ≥65 y, n (%) | 2 (29) | 5 (42) | 5 (56) | 4 (44) | 5 (63) | 10 (48) | 21 (47) |
| Male sex, n (%) | 4 (57) | 8 (67) | 4 (44) | 4 (44) | 5 (63) | 12 (57) | 25 (56) |
| White race, n (%)* | 5 (71) | 11 (92) | 8 (89) | 9 (100) | 7 (88) | 19 (90) | 40 (89) |
| Mean BMI, kg/m2 (SD) | 29.0 (5.80) | 31.0 (10.30) | 29.3 (5.10) | 28.0 (5.50) | 25.9 (4.49) | 30.3 (8.33) | 28.8 (6.90) |
| ECOG PS, n (%) | |||||||
| 0 | 1 (14) | 7 (58) | 2 (22) | 3 (33) | 1 (13) | 9 (43) | 14 (31) |
| 1 | 6 (86) | 5 (42) | 7 (78) | 6 (67) | 7 (88) | 12 (57) | 31 (69) |
| Median time from initial diagnosis, mo (range) | 14.4 (6-38) | 15.2 (8-83) | 11.2 (3-252) | 11.8 (9-72) | 16.9 (7-154) | 14.1 (3-252) | 14.1 (3-252) |
| DLBCL subtype, n (%)† | |||||||
| Non-GCB | 2 (29) | 5 (42) | 8 (89) | 5 (56) | 3 (37) | 13 (61) | 23 (51) |
| GCB | 3 (42) | 5 (42) | 1 (11) | 3 (33) | 5 (63) | 6 (29) | 17 (38) |
| Not reported/missing | 2 (29) | 2 (16) | 0 (0) | 1 (11) | 0 (0) | 2 (10) | 5 (11) |
| DLBCL category, n (%) | |||||||
| De novo | 5 (71) | 8 (67) | 7 (78) | 6 (67) | 4 (50) | 15 (71) | 30 (67) |
| Transformed | 2 (29) | 4 (33) | 2 (22) | 3 (33) | 4 (50) | 6 (29) | 15 (33) |
| Ann Arbor staging, n (%) | |||||||
| II/IIE | 3 (42) | 1 (8) | 1 (11) | 1 (11) | 0 (0) | 2 (9) | 6 (13) |
| III/IIIE/IIIE,S | 2 (29) | 1 (8) | 0 (0) | 2 (22) | 3 (37) | 1 (5) | 8 (18) |
| IV | 2 (29) | 10 (84) | 8 (89) | 6 (67) | 5 (63) | 18 (86) | 31 (69) |
| Bulky disease, n (%) | 6 (86) | 5 (42) | 3 (33) | 5 (56) | 5 (63) | 8 (38) | 24 (53) |
| 5-10 cm | 4 (57) | 2 (17) | 2 (22) | 3 (33) | 5 (63) | 4 (19) | 16 (36) |
| >10 cm | 2 (29) | 3 (25) | 1 (11) | 2 (22) | 0 (0) | 4 (19) | 8 (18) |
| No. of prior regimens, n (%) | |||||||
| 1 | 0 (0) | 2 (17) | 2 (22) | 2 (22) | 1 (13) | 4 (19) | 7 (16) |
| ≥2 | 7 (100) | 10 (83) | 7 (78) | 7 (78) | 7 (88) | 17 (81) | 38 (84) |
| Disease status at completion of treatment regimen preceding study entry, n (%) | |||||||
| Refractory‡ | 4 (57) | 8 (67) | 5 (56) | 5 (56) | 5 (63) | 13 (62) | 27 (60) |
| Relapsed | 3 (43) | 4 (33) | 4 (44) | 4 (44) | 3 (38) | 8 (38) | 18 (40) |
| CR | 1 (14) | 3 (25) | 4 (44) | 4 (44) | 3 (38) | 7 (33) | 15 (33) |
| PR | 2 (29) | 1 (8) | 0 (0) | 0 (0) | 0 (0) | 1 (5) | 3 (7) |
| Primary refractory disease, n (%)¶ | 5 (71) | 3 (25) | 2 (22) | 1 (11) | 1 (13) | 5 (24) | 12 (27) |
| Prior autologous transplant§ | 2 (29) | 3 (25) | 4 (44) | 0 (0) | 2 (25) | 7 (33) | 11 (24) |
| Median time from last dose of prior therapy to first ibrutinib dose, mo (range) | 1.9 (1.1-20.7) | 2.2 (0.7-20.6) | 2.4 (0.7-36.6) | 2.5 (2.1-68.3) | 2.7 (0.0-65.7) | 2.4 (0.7-36.6) | 2.4 (0.0-68.3) |
| 560 mg/day ibrutinib + 375 mg/m2 IV rituximab combined with: | |||||||
|---|---|---|---|---|---|---|---|
| Dose level −1: 10 mg LEN (n = 7) | Dose level 1: 15 mg LEN (n = 12) | Dose level 1+: 15 mg LEN (n = 9) | Dose level 2: 20 mg LEN (n = 9) | Dose level 3: 25 mg LEN (n = 8) | Dose level 1 combined (n = 21) | All dose levels (N = 45) | |
| Age | |||||||
| Median age, y (range) | 59 (49-68) | 62 (43-84) | 68 (41-85) | 64 (50-77) | 68 (36-78) | 64 (41-85) | 64 (36-85) |
| ≥65 y, n (%) | 2 (29) | 5 (42) | 5 (56) | 4 (44) | 5 (63) | 10 (48) | 21 (47) |
| Male sex, n (%) | 4 (57) | 8 (67) | 4 (44) | 4 (44) | 5 (63) | 12 (57) | 25 (56) |
| White race, n (%)* | 5 (71) | 11 (92) | 8 (89) | 9 (100) | 7 (88) | 19 (90) | 40 (89) |
| Mean BMI, kg/m2 (SD) | 29.0 (5.80) | 31.0 (10.30) | 29.3 (5.10) | 28.0 (5.50) | 25.9 (4.49) | 30.3 (8.33) | 28.8 (6.90) |
| ECOG PS, n (%) | |||||||
| 0 | 1 (14) | 7 (58) | 2 (22) | 3 (33) | 1 (13) | 9 (43) | 14 (31) |
| 1 | 6 (86) | 5 (42) | 7 (78) | 6 (67) | 7 (88) | 12 (57) | 31 (69) |
| Median time from initial diagnosis, mo (range) | 14.4 (6-38) | 15.2 (8-83) | 11.2 (3-252) | 11.8 (9-72) | 16.9 (7-154) | 14.1 (3-252) | 14.1 (3-252) |
| DLBCL subtype, n (%)† | |||||||
| Non-GCB | 2 (29) | 5 (42) | 8 (89) | 5 (56) | 3 (37) | 13 (61) | 23 (51) |
| GCB | 3 (42) | 5 (42) | 1 (11) | 3 (33) | 5 (63) | 6 (29) | 17 (38) |
| Not reported/missing | 2 (29) | 2 (16) | 0 (0) | 1 (11) | 0 (0) | 2 (10) | 5 (11) |
| DLBCL category, n (%) | |||||||
| De novo | 5 (71) | 8 (67) | 7 (78) | 6 (67) | 4 (50) | 15 (71) | 30 (67) |
| Transformed | 2 (29) | 4 (33) | 2 (22) | 3 (33) | 4 (50) | 6 (29) | 15 (33) |
| Ann Arbor staging, n (%) | |||||||
| II/IIE | 3 (42) | 1 (8) | 1 (11) | 1 (11) | 0 (0) | 2 (9) | 6 (13) |
| III/IIIE/IIIE,S | 2 (29) | 1 (8) | 0 (0) | 2 (22) | 3 (37) | 1 (5) | 8 (18) |
| IV | 2 (29) | 10 (84) | 8 (89) | 6 (67) | 5 (63) | 18 (86) | 31 (69) |
| Bulky disease, n (%) | 6 (86) | 5 (42) | 3 (33) | 5 (56) | 5 (63) | 8 (38) | 24 (53) |
| 5-10 cm | 4 (57) | 2 (17) | 2 (22) | 3 (33) | 5 (63) | 4 (19) | 16 (36) |
| >10 cm | 2 (29) | 3 (25) | 1 (11) | 2 (22) | 0 (0) | 4 (19) | 8 (18) |
| No. of prior regimens, n (%) | |||||||
| 1 | 0 (0) | 2 (17) | 2 (22) | 2 (22) | 1 (13) | 4 (19) | 7 (16) |
| ≥2 | 7 (100) | 10 (83) | 7 (78) | 7 (78) | 7 (88) | 17 (81) | 38 (84) |
| Disease status at completion of treatment regimen preceding study entry, n (%) | |||||||
| Refractory‡ | 4 (57) | 8 (67) | 5 (56) | 5 (56) | 5 (63) | 13 (62) | 27 (60) |
| Relapsed | 3 (43) | 4 (33) | 4 (44) | 4 (44) | 3 (38) | 8 (38) | 18 (40) |
| CR | 1 (14) | 3 (25) | 4 (44) | 4 (44) | 3 (38) | 7 (33) | 15 (33) |
| PR | 2 (29) | 1 (8) | 0 (0) | 0 (0) | 0 (0) | 1 (5) | 3 (7) |
| Primary refractory disease, n (%)¶ | 5 (71) | 3 (25) | 2 (22) | 1 (11) | 1 (13) | 5 (24) | 12 (27) |
| Prior autologous transplant§ | 2 (29) | 3 (25) | 4 (44) | 0 (0) | 2 (25) | 7 (33) | 11 (24) |
| Median time from last dose of prior therapy to first ibrutinib dose, mo (range) | 1.9 (1.1-20.7) | 2.2 (0.7-20.6) | 2.4 (0.7-36.6) | 2.5 (2.1-68.3) | 2.7 (0.0-65.7) | 2.4 (0.7-36.6) | 2.4 (0.0-68.3) |
BMI, body mass index; ECOG PS, Eastern Cooperative Oncology Group performance status; SD, standard deviation.
1 patient in dose level −1 had unknown race.
Based on local pathology laboratory reports.
Defined as stable disease or progressive disease at completion of treatment regimen preceding study entry.
Defined as refractory to front-line regimen (only received 1 prior therapy) based on medical history.
Two patients with prior autologous transplant did not respond to salvage therapy.
Median follow-up time was 25.6 months (range, 0.4–44.8 months). As of this analysis, 6 (13%) patients were still receiving ibrutinib and lenalidomide (time on treatment, 19.8-45.0 months; Figure 1B). Most (n = 25 [56%]) patients received at least 3 months of study drug, including 2 (4%) who received from 12 or more to less than 18 months of treatment and 7 (16%) who received at least 18 months of treatment (Table 2). Sixteen (36%) patients completed the first 6 cycles, which included 6 rituximab doses. Of 39 (87%) patients who discontinued study drug early, most (n = 29) discontinued because of PD. Deaths were reported for 28 (62%) patients, including 6 (13%) who died receiving treatment or within 30 days of treatment (because of PD, n = 3; because of an AE, n = 3 [cardiac arrest, pneumonia, Escherichia sepsis]). The patient with cardiac arrest was an 84-year-old woman who received study treatment for 104 days before discontinuing; cardiac arrest occurred 8 days after initiation of subsequent anticancer therapy, with death occurring 20 days after study treatment discontinuation, and was considered unrelated to study treatment. The patient with pneumonia was a 68-year-old man who received study treatment for 168 days before discontinuing; the patient initiated subsequent anticancer therapy 3 days later, and in the following 7 to 11 days, developed grade 4 thrombocytopenia, grade 3 dehydration, grade 4 Pseudomonas bacterial infection, and grade 3 left ventricular dysfunction, with death occurring 19 days after study treatment discontinuation. The patient with Escherichia sepsis was an 85-year-old man who received study treatment for 30 days before a temporary interruption while traveling; the patient had ongoing grade 2 thrombocytopenia since day 8 and grade 2 anemia since day 15 and had a normal neutrophil count. Sepsis was first noted on day 33, and death occurred on day 34.
Treatment exposure and dose modifications
| 560 mg/day ibrutinib + 375 mg/m2 IV rituximab combined with: | |||||||
|---|---|---|---|---|---|---|---|
| Dose level −1:10 mg LEN (n = 7) | Dose level 1:15 mg LEN (n = 12) | Dose level 1+:15 mg LEN (n = 9) | Dose level 2:20 mg LEN (n = 9) | Dose level 3:25 mg LEN (n = 8) | Dose level 1: combined (n = 21) | All dose levels (N = 45) | |
| No. of treatment cycles received, n (%) | |||||||
| 1 | 2 (29) | 3 (25) | 1 (11) | 1 (11) | 1 (13) | 4 (19) | 8 (18) |
| 2 | 2 (29) | 1 (8) | 1 (11) | 2 (22) | 0 (0) | 2 (10) | 6 (13) |
| 3-5 | 0 (0) | 4 (33) | 2 (22) | 4 (44) | 1 (13) | 6 (29) | 11 (24) |
| 6 | 1 (14) | 0 (0) | 1 (11) | 1 (11) | 2 (25) | 1 (5) | 5 (11) |
| 7-12 | 2 (29) | 2 (17) | 1 (11) | 0 (0) | 1 (13) | 3 (14) | 6 (13) |
| >12 | 0 (0) | 2 (17) | 3 (33) | 1 (11) | 3 (38) | 5 (24) | 9 (20) |
| Mean relative dose intensity, % (SD)* | |||||||
| Ibrutinib | 94.6 (6.50) | 89.1 (14.11) | 80.5 (13.44) | 85.3 (18.98) | 78.3 (21.44) | 85.4 (14.17) | 85.6 (16.07) |
| Lenalidomide | 79.6 (34.30) | 81.2 (16.48) | 69.5 (27.02) | 69.6 (27.70) | 64.9 (26.45) | 76.2 (21.83) | 73.4 (25.57) |
| Rituximab | 100 (0.00) | 100 (0.00) | 100 (0.00) | 98.6 (4.17) | 100.1 (0.35) | 100 (0.00) | 99.7 (1.87) |
| Dose delay or reduction because of an AE, n (%) | 4 (57) | 7 (58) | 7 (78) | 8 (89) | 8 (100) | 14 (67) | 34 (76) |
| Dose reduction† | 0 (0) | 4 (33) | 4 (44) | 4 (44) | 5 (63) | 8 (38) | 17 (38) |
| Ibrutinib | 0 (0) | 2 (17) | 4 (44) | 3 (33) | 3 (38) | 6 (29) | 12 (27) |
| Lenalidomide | 0 (0) | 2 (17) | 3 (33) | 4 (44) | 4 (50) | 5 (24) | 13 (29) |
| Rituximab | 0 (0) | 0 (0) | 0 (0) | 1 (11) | 0 (0) | 0 (0) | 1 (2) |
| Dose delay‡ | 4 (57) | 7 (58) | 7 (78) | 8 (89) | 8 (100) | 14 (67) | 34 (76) |
| Ibrutinib | 4 (57) | 6 (50) | 7 (78) | 8 (89) | 8 (100) | 13 (62) | 33 (73) |
| Lenalidomide | 2 (29) | 4 (33) | 4 (44) | 7 (78) | 7 (88) | 8 (38) | 24 (53) |
| Rituximab | 1 (14) | 1 (8) | 1 (11) | 0 (0) | 0 (0) | 2 (10) | 3 (7) |
| 560 mg/day ibrutinib + 375 mg/m2 IV rituximab combined with: | |||||||
|---|---|---|---|---|---|---|---|
| Dose level −1:10 mg LEN (n = 7) | Dose level 1:15 mg LEN (n = 12) | Dose level 1+:15 mg LEN (n = 9) | Dose level 2:20 mg LEN (n = 9) | Dose level 3:25 mg LEN (n = 8) | Dose level 1: combined (n = 21) | All dose levels (N = 45) | |
| No. of treatment cycles received, n (%) | |||||||
| 1 | 2 (29) | 3 (25) | 1 (11) | 1 (11) | 1 (13) | 4 (19) | 8 (18) |
| 2 | 2 (29) | 1 (8) | 1 (11) | 2 (22) | 0 (0) | 2 (10) | 6 (13) |
| 3-5 | 0 (0) | 4 (33) | 2 (22) | 4 (44) | 1 (13) | 6 (29) | 11 (24) |
| 6 | 1 (14) | 0 (0) | 1 (11) | 1 (11) | 2 (25) | 1 (5) | 5 (11) |
| 7-12 | 2 (29) | 2 (17) | 1 (11) | 0 (0) | 1 (13) | 3 (14) | 6 (13) |
| >12 | 0 (0) | 2 (17) | 3 (33) | 1 (11) | 3 (38) | 5 (24) | 9 (20) |
| Mean relative dose intensity, % (SD)* | |||||||
| Ibrutinib | 94.6 (6.50) | 89.1 (14.11) | 80.5 (13.44) | 85.3 (18.98) | 78.3 (21.44) | 85.4 (14.17) | 85.6 (16.07) |
| Lenalidomide | 79.6 (34.30) | 81.2 (16.48) | 69.5 (27.02) | 69.6 (27.70) | 64.9 (26.45) | 76.2 (21.83) | 73.4 (25.57) |
| Rituximab | 100 (0.00) | 100 (0.00) | 100 (0.00) | 98.6 (4.17) | 100.1 (0.35) | 100 (0.00) | 99.7 (1.87) |
| Dose delay or reduction because of an AE, n (%) | 4 (57) | 7 (58) | 7 (78) | 8 (89) | 8 (100) | 14 (67) | 34 (76) |
| Dose reduction† | 0 (0) | 4 (33) | 4 (44) | 4 (44) | 5 (63) | 8 (38) | 17 (38) |
| Ibrutinib | 0 (0) | 2 (17) | 4 (44) | 3 (33) | 3 (38) | 6 (29) | 12 (27) |
| Lenalidomide | 0 (0) | 2 (17) | 3 (33) | 4 (44) | 4 (50) | 5 (24) | 13 (29) |
| Rituximab | 0 (0) | 0 (0) | 0 (0) | 1 (11) | 0 (0) | 0 (0) | 1 (2) |
| Dose delay‡ | 4 (57) | 7 (58) | 7 (78) | 8 (89) | 8 (100) | 14 (67) | 34 (76) |
| Ibrutinib | 4 (57) | 6 (50) | 7 (78) | 8 (89) | 8 (100) | 13 (62) | 33 (73) |
| Lenalidomide | 2 (29) | 4 (33) | 4 (44) | 7 (78) | 7 (88) | 8 (38) | 24 (53) |
| Rituximab | 1 (14) | 1 (8) | 1 (11) | 0 (0) | 0 (0) | 2 (10) | 3 (7) |
Calculated as the total received dose level divided by the total expected dose level up to the last treatment.
Ibrutinib dose reduced to 420 mg in 11 (24%) patients and 280 mg in 4 (9%) patients; lenalidomide dose reduced to 20 mg in 3 (7%) patients, 15 mg in 6 (13%) patients, 10 mg in 11 (24%) patients, and 5 mg in 5 (11%) patients; rituximab dose reduced to 450 mg in 1 (2%) patient.
Delays of ≥7 d reported for ibrutinib in 23 (51%) patients and for lenalidomide in 15 (33%) patients.
Safety and tolerability
Cohort 1 enrolled 12 patients treated with ibrutinib, rituximab, and 15 mg lenalidomide. Three patients in this cohort experienced DLTs, including grade 3 rash (n = 2) and grade 3 neutropenia lasting longer than 7 days (n = 1); the 3 DLTs required enrollment of a de-escalation cohort (cohort −1; 10-mg lenalidomide dose; Table 3). One of 7 patients in cohort −1 experienced a DLT (grade 4 Salmonella sepsis), and re-escalation to the 15-mg lenalidomide dose subsequently occurred (cohort 1+), with additional toxicity management guidelines used as described in the supplemental Methods, available on the Blood Web site. Nine patients were treated in cohort 1+ with 1 DLT of grade 4 neutropenia. Thus, 21 patients were treated in cohorts 1 and 1+ at the 15-mg lenalidomide dose, but enrollment was separated by several months. Escalation to cohort 2 (n = 9; 20-mg lenalidomide dose) and cohort 3 (n = 8; 25-mg lenalidomide dose) subsequently occurred with no DLTs; therefore, the maximum tolerated dose was not reached.
Summary of dose-escalation cohorts, lenalidomide dose levels, and DLTs
| Cohort | Enrollment | Description | Lenalidomide dose level* | DLTs |
|---|---|---|---|---|
| 1 | n = 12 | Starting dose | 15 mg | • Grade 3 neutropenia lasting >7 d (n = 2) • Grade 3 rash (n = 2) |
| −1 | n = 7 | De-escalation dose | 10 mg | • Grade 4 Salmonella sepsis (n = 1) |
| Protocol amendment to relax criteria for DLTs of neutropenia and rash, and to include management guidelines for toxicities | ||||
| 1+ | n = 9 | Re-escalation dose | 15 mg | • Grade 4 neutropenia (n = 1) |
| 2 | n = 9 | Phase 2 dose | 20 mg | None |
| 3 | n = 8 | Maximum dose level tested | 25 mg | None |
| Cohort | Enrollment | Description | Lenalidomide dose level* | DLTs |
|---|---|---|---|---|
| 1 | n = 12 | Starting dose | 15 mg | • Grade 3 neutropenia lasting >7 d (n = 2) • Grade 3 rash (n = 2) |
| −1 | n = 7 | De-escalation dose | 10 mg | • Grade 4 Salmonella sepsis (n = 1) |
| Protocol amendment to relax criteria for DLTs of neutropenia and rash, and to include management guidelines for toxicities | ||||
| 1+ | n = 9 | Re-escalation dose | 15 mg | • Grade 4 neutropenia (n = 1) |
| 2 | n = 9 | Phase 2 dose | 20 mg | None |
| 3 | n = 8 | Maximum dose level tested | 25 mg | None |
DLT, dose-limiting toxicity.
Lenalidomide dose in combination with 560 mg/day ibrutinib and 375 mg/m2 intravenous rituximab.
The most frequently reported TEAEs across cohorts were gastrointestinal events, myelosuppression, fatigue, hypokalemia, peripheral edema, and maculopapular rash (Table 4). Five patients experienced new-onset atrial fibrillation (AF); none had a history of AF. Two patients had prior ongoing AF at enrollment, and neither experienced worsening of AF during study treatment. None of the patients who experienced AF on study discontinued study drugs because of AF. Most patients (n = 42 [93%]) experienced a grade 3 or worse TEAE, including 34 (76%) who experienced a grade 3 or worse event considered related to the study drug. The most frequent grade 3 or 4 TEAEs related to ibrutinib or lenalidomide were neutropenia (n = 17 [38%] for ibrutinib and n = 18 [40%] for lenalidomide), maculopapular rash (n = 6 [13%] for both), and thrombocytopenia (n = 4 [9%] for ibrutinib and n = 5 [11%] for lenalidomide). Serious TEAEs were reported for 25 (56%) patients, including 11 (24%) with a treatment-related serious TEAE. The most frequent serious TEAEs were worsening of DLBCL (n = 5 [11%]), febrile neutropenia, AF, and dehydration (n = 3 [7%] each; Table 4). Of 3 patients with serious TEAEs of febrile neutropenia (all grade 3), 2 had events occurring on days 154 to 156 and days 183 to 189 that were considered unrelated to study treatment; the third patient had events on days 236 to 240 (with concurrent grade 3 atrial fibrillation) and days 436 to 440 that were considered related to ibrutinib and lenalidomide. AF was considered related to study treatment in all 3 patients. One patient experienced grade 2 AF on days 23 to 25, another experienced grade 3 AF on days 60 to 61 (with concurrent grade 3 dehydration) and days 88 to 91, and a third experienced grade 3 AF on days 125 to 133, days 169 to 172, and days 236 to 240 (with concurrent grade 3 febrile neutropenia). Patients with febrile neutropenia and AF were typically treated and hospitalized as clinically indicated, and study medication was frequently delayed and/or reduced (supplemental Table 1, available on the Blood Web site). Dehydration was considered unrelated to study treatment and resolved after 1 to 3 days in all 3 cases. Five patients experienced grade 5 TEAEs, including Escherichia sepsis (n = 1; see "Patients") and worsening of DLBCL (n = 4).
Summary of TEAEs
| 560 mg/day ibrutinib + 375 mg/m2 IV rituximab combined with: | |||||||
|---|---|---|---|---|---|---|---|
| Dose level −1: 10 mg LEN (n = 7) | Dose level 1: 15 mg LEN (n = 12) | Dose level 1+:15 mg LEN (n = 9) | Dose level 2:20 mg LEN (n = 9) | Dose level 3:25 mg LEN (n = 8) | Dose level 1 combined (n = 21) | All dose levels (N = 45) | |
| TEAEs in ≥20% of patients, n (%) | |||||||
| Diarrhea | 3 (43) | 9 (75) | 2 (22) | 4 (44) | 6 (75) | 11 (52) | 24 (53) |
| Nausea | 3 (43) | 7 (58) | 4 (44) | 3 (33) | 5 (63) | 11 (52) | 22 (49) |
| Fatigue | 3 (43) | 6 (50) | 5 (56) | 2 (22) | 4 (50) | 11 (52) | 20 (44) |
| Neutropenia | 0 (0) | 5 (42) | 6 (67) | 3 (33) | 4 (50) | 11 (52) | 18 (40) |
| Constipation | 2 (29) | 5 (42) | 4 (44) | 4 (44) | 2 (25) | 9 (43) | 17 (38) |
| Thrombocytopenia | 2 (29) | 4 (33) | 3 (33) | 4 (44) | 3 (38) | 7 (33) | 16 (36) |
| Hypokalemia | 1 (14) | 4 (33) | 3 (33) | 3 (33) | 3 (38) | 7 (33) | 14 (31) |
| Anemia | 0 (0) | 3 (25) | 5 (56) | 3 (33) | 2 (25) | 8 (38) | 13 (29) |
| Peripheral edema | 1 (14) | 2 (17) | 3 (33) | 3 (33) | 4 (50) | 5 (24) | 13 (29) |
| Maculopapular rash | 1 (14) | 2 (17) | 5 (56) | 3 (33) | 2 (25) | 7 (33) | 13 (29) |
| Vomiting | 1 (14) | 3 (25) | 2 (22) | 3 (33) | 3 (38) | 5 (24) | 12 (27) |
| Decreased appetite | 1 (14) | 2 (17) | 4 (44) | 2 (22) | 2 (25) | 6 (29) | 11 (24) |
| Dizziness | 0 (0) | 4 (33) | 2 (22) | 3 (33) | 2 (25) | 6 (29) | 11 (24) |
| Dyspnea | 1 (14) | 3 (25) | 1 (11) | 3 (33) | 2 (25) | 4 (19) | 10 (22) |
| Abdominal pain | 2 (29) | 2 (17) | 0 (0) | 3 (33) | 2 (25) | 2 (10) | 9 (20) |
| Decreased weight | 2 (29) | 3 (25) | 0 (0) | 1 (11) | 3 (38) | 3 (14) | 9 (20) |
| Any grade 3-4 TEAE, n (%) | 5 (71) | 11 (92) | 8 (89) | 9 (100) | 7 (88) | 19 (90) | 40 (89) |
| Ibrutinib related | 3 (43) | 10 (83) | 6 (67) | 7 (78) | 6 (75) | 16 (76) | 32 (71) |
| Lenalidomide related | 3 (43) | 9 (75) | 7 (78) | 7 (78) | 5 (63) | 16 (76) | 31 (69) |
| Any serious TEAE, n (%) | 5 (71) | 3 (25) | 7 (78) | 5 (56) | 5 (63) | 10 (48) | 25 (56) |
| Serious TEAEs in ≥5% of patients, n (%) | |||||||
| Worsening of DLBCL | 1 (14) | 0 (0) | 1 (11) | 2 (22) | 1 (13) | 1 (5) | 5 (11) |
| Febrile neutropenia | 0 (0) | 0 (0) | 0 (0) | 2 (22) | 1 (13) | 0 (0) | 3 (7) |
| Atrial fibrillation | 0 (0) | 0 (0) | 0 (0) | 2 (22) | 1 (13) | 0 (0) | 3 (7) |
| Dehydration | 0 (0) | 0 (0) | 1 (11) | 1 (11) | 1 (13) | 1 (5) | 3 (7) |
| Any grade 5 TEAE, n (%)* | 1 (14) | 0 (0) | 2 (22) | 1 (11) | 1 (13) | 2 (10) | 5 (11) |
| Discontinuation due to TEAE, n (%) | 1 (14) | 4 (33) | 3 (33) | 2 (22) | 1 (13) | 7 (33) | 11 (24) |
| 560 mg/day ibrutinib + 375 mg/m2 IV rituximab combined with: | |||||||
|---|---|---|---|---|---|---|---|
| Dose level −1: 10 mg LEN (n = 7) | Dose level 1: 15 mg LEN (n = 12) | Dose level 1+:15 mg LEN (n = 9) | Dose level 2:20 mg LEN (n = 9) | Dose level 3:25 mg LEN (n = 8) | Dose level 1 combined (n = 21) | All dose levels (N = 45) | |
| TEAEs in ≥20% of patients, n (%) | |||||||
| Diarrhea | 3 (43) | 9 (75) | 2 (22) | 4 (44) | 6 (75) | 11 (52) | 24 (53) |
| Nausea | 3 (43) | 7 (58) | 4 (44) | 3 (33) | 5 (63) | 11 (52) | 22 (49) |
| Fatigue | 3 (43) | 6 (50) | 5 (56) | 2 (22) | 4 (50) | 11 (52) | 20 (44) |
| Neutropenia | 0 (0) | 5 (42) | 6 (67) | 3 (33) | 4 (50) | 11 (52) | 18 (40) |
| Constipation | 2 (29) | 5 (42) | 4 (44) | 4 (44) | 2 (25) | 9 (43) | 17 (38) |
| Thrombocytopenia | 2 (29) | 4 (33) | 3 (33) | 4 (44) | 3 (38) | 7 (33) | 16 (36) |
| Hypokalemia | 1 (14) | 4 (33) | 3 (33) | 3 (33) | 3 (38) | 7 (33) | 14 (31) |
| Anemia | 0 (0) | 3 (25) | 5 (56) | 3 (33) | 2 (25) | 8 (38) | 13 (29) |
| Peripheral edema | 1 (14) | 2 (17) | 3 (33) | 3 (33) | 4 (50) | 5 (24) | 13 (29) |
| Maculopapular rash | 1 (14) | 2 (17) | 5 (56) | 3 (33) | 2 (25) | 7 (33) | 13 (29) |
| Vomiting | 1 (14) | 3 (25) | 2 (22) | 3 (33) | 3 (38) | 5 (24) | 12 (27) |
| Decreased appetite | 1 (14) | 2 (17) | 4 (44) | 2 (22) | 2 (25) | 6 (29) | 11 (24) |
| Dizziness | 0 (0) | 4 (33) | 2 (22) | 3 (33) | 2 (25) | 6 (29) | 11 (24) |
| Dyspnea | 1 (14) | 3 (25) | 1 (11) | 3 (33) | 2 (25) | 4 (19) | 10 (22) |
| Abdominal pain | 2 (29) | 2 (17) | 0 (0) | 3 (33) | 2 (25) | 2 (10) | 9 (20) |
| Decreased weight | 2 (29) | 3 (25) | 0 (0) | 1 (11) | 3 (38) | 3 (14) | 9 (20) |
| Any grade 3-4 TEAE, n (%) | 5 (71) | 11 (92) | 8 (89) | 9 (100) | 7 (88) | 19 (90) | 40 (89) |
| Ibrutinib related | 3 (43) | 10 (83) | 6 (67) | 7 (78) | 6 (75) | 16 (76) | 32 (71) |
| Lenalidomide related | 3 (43) | 9 (75) | 7 (78) | 7 (78) | 5 (63) | 16 (76) | 31 (69) |
| Any serious TEAE, n (%) | 5 (71) | 3 (25) | 7 (78) | 5 (56) | 5 (63) | 10 (48) | 25 (56) |
| Serious TEAEs in ≥5% of patients, n (%) | |||||||
| Worsening of DLBCL | 1 (14) | 0 (0) | 1 (11) | 2 (22) | 1 (13) | 1 (5) | 5 (11) |
| Febrile neutropenia | 0 (0) | 0 (0) | 0 (0) | 2 (22) | 1 (13) | 0 (0) | 3 (7) |
| Atrial fibrillation | 0 (0) | 0 (0) | 0 (0) | 2 (22) | 1 (13) | 0 (0) | 3 (7) |
| Dehydration | 0 (0) | 0 (0) | 1 (11) | 1 (11) | 1 (13) | 1 (5) | 3 (7) |
| Any grade 5 TEAE, n (%)* | 1 (14) | 0 (0) | 2 (22) | 1 (11) | 1 (13) | 2 (10) | 5 (11) |
| Discontinuation due to TEAE, n (%) | 1 (14) | 4 (33) | 3 (33) | 2 (22) | 1 (13) | 7 (33) | 11 (24) |
Includes study treatment–related Escherichia sepsis in 1 patient in dose cohort 1+ and worsening of DLBCL in 4 patients. One additional death occurred that was not considered a TEAE, as it occurred 16 days after receiving subsequent anticancer therapy in a patient who discontinued ibrutinib. TEAEs were defined as AEs occurring after the first dose of study drugs and within 30 days after the last dose of study drug; any AE that is considered study drug-related regardless of the start date of the event; or any AE that is present at baseline but worsens after the first administration of study drug in severity or is subsequently considered drug-related by the investigator.
Eleven patients discontinued treatment because of a TEAE (cohort 1, n = 4 [maculopapular rash, n = 2; neutropenia, n = 1; intraductal proliferative breast lesion, n = 1]; cohort −1, n = 1 [Salmonella sepsis]; cohort 1+, n = 3 [Escherichia sepsis, worsening of DLBCL, ocular lymphoma]; cohort 2, n = 2 [both worsening of DLBCL]; cohort 3, n = 1 [worsening of DLBCL]). Dose delays and/or reductions resulting from AEs occurred more frequently in cohorts 1+, 2, and 3 (ibrutinib, 78%-100%; lenalidomide, 44%-100%) than in cohort 1 (ibrutinib, 50%; lenalidomide, 42%; Table 2).
Activity
Across all dose levels (n = 45), ORR was 38% (95% confidence interval [CI], 24%-54%) and included 11 (24%) patients who achieved CR (confirmed by positron emission tomography scan). Seven (16%) patients achieved stable disease (3/7 with primary refractory disease), and 16 (36%) had PD. Five patients (11%; all with non-GCB DLBCL and treated with 15 mg lenalidomide [cohort 1, n = 3; cohort 1+, n = 2]) were nonevaluable for response (a sixth patient died of DLBCL after 5 days of treatment). In response-evaluable patients (n = 39), ORR was 44% (95% CI, 28%-60%; Figure 2A). As expected, responses were more common among patients with non-GCB (n = 17; 65% [95% CI, 38%-86%], including 41% with CR) compared with GCB DLBCL (n = 17; 29% [95% CI, 10%-56%], including 18% with CR; Figure 2B; supplemental Table 2). Among 7 response-evaluable patients with the ABC phenotype, as determined by gene expression profiling, 2 patients achieved CR and 1 patient achieved PR, for an ORR of 43% (tumor sample availability limited gene expression profiling). There were three patients with double-hit DLBCL (1 CR, 2 PD). Among response-evaluable patients, ORR was 26% (95% CI, 10%-48%) in those with refractory disease (n = 23), including 3 (13%) patients with CR; ORR was 17% and 56% in patients with primary (n = 12) and secondary (n = 27) refractory disease, respectively. ORR was 69% in patients with relapsed disease (n = 16), and 50% of patients achieved CR. Among 24 patients with bulky disease, 2 achieved CR and 4 achieved PR (Figure 3). ORR and CR rate for 13 response-evaluable patients with transformed disease were 46% and 15%, respectively (Figure 2B). For responders, the median duration of response (DOR) as of the date of this analysis was 15.9 months (range, 0.9-37.2+ months) for all patients, 15.9 months (range, 0.9-36.5+ months) for patients with non-GCB DLBCL, 8.8 months (range, 1.8-14.0+ months) for patients with GCB DLBCL, and 12.3 months (range, 2.8-37.2+ months) for patients with transformed DLBCL; however, several patients were still receiving study treatment (Figure 1). Among patients who achieved CR, the median DOR has not been reached (range, 1.8-37.2+ months).
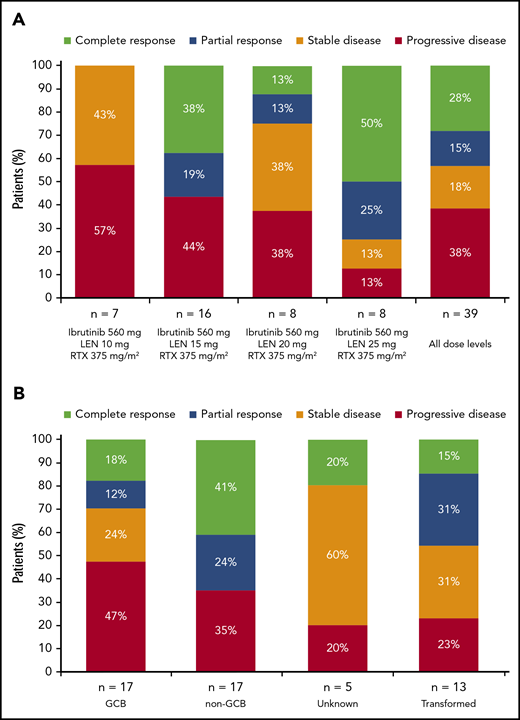
Best response. Data are presented for patients in the response-evaluable population by dose cohort (A) and by DLBCL subtype/category (B). Five patients with non-GCB DLBCL treated with 15 mg lenalidomide (cohort 1, n = 3; cohort 1+, n = 2) were nonevaluable for response, and a sixth patient (20 mg lenalidomide) died of DLBCL after 5 days of treatment. The response-evaluable population included patients who had measurable disease at baseline and at least 1 posttreatment disease assessment by the investigator. LEN, lenalidomide; RTX, rituximab.
Best response. Data are presented for patients in the response-evaluable population by dose cohort (A) and by DLBCL subtype/category (B). Five patients with non-GCB DLBCL treated with 15 mg lenalidomide (cohort 1, n = 3; cohort 1+, n = 2) were nonevaluable for response, and a sixth patient (20 mg lenalidomide) died of DLBCL after 5 days of treatment. The response-evaluable population included patients who had measurable disease at baseline and at least 1 posttreatment disease assessment by the investigator. LEN, lenalidomide; RTX, rituximab.
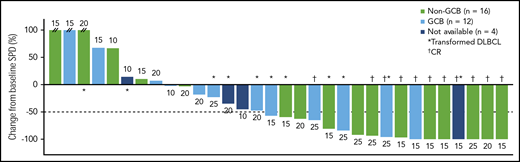
Maximum percentage reduction in tumor size. Data are presented for patients in the all-treated population who had a baseline and at least 1 postbaseline tumor assessment. The number next to each bar indicates the dose cohort (ie, lenalidomide dose) for that patient. // indicates values greater than 100% (112%, 153%, and 414%). SPD, sum of the product of the diameters.
Maximum percentage reduction in tumor size. Data are presented for patients in the all-treated population who had a baseline and at least 1 postbaseline tumor assessment. The number next to each bar indicates the dose cohort (ie, lenalidomide dose) for that patient. // indicates values greater than 100% (112%, 153%, and 414%). SPD, sum of the product of the diameters.
Median progression-free survival (PFS) among all patients was 5.5 months (95% CI, 2.8-6.4 months), and for responders it was 18.8 months (95% CI, 5.8 months-not estimable [NE]). Median OS was 9.5 months (95% CI, 6.5-29.2 months) among all patients and not reached (95% CI, 11.6 months-NE) among responders. Among patients with non-GCB DLBCL, median PFS was 3.9 months (95% CI, 2.1-15.1 months) and median OS was 10.7 months (95% CI, 6.1 months-NE). Among patients with GCB DLBCL, median PFS was 5.5 months (95% CI, 1.4-5.9 months) and median OS was 7.1 months (95% CI, 3.4-29.2 months). Among patients with transformed DLBCL, median PFS was 5.9 months (95% CI, 1.6-11.6 months) and median OS was 12.0 months (95% CI, 3.3 months-NE).
Discussion
This initial clinical evaluation of ibrutinib, lenalidomide, and rituximab in adults with relapsed/refractory DLBCL identified a recommended phase 2 dose of 560 mg ibrutinib, 375 mg/m2 IV rituximab daily on Day 1 of Cycles 1-6, and 20 mg lenalidomide on Days 1-21 of each 28-day cycle. Based on efficacy and safety considerations and the desire to start phase 2, the 20-mg lenalidomide dose was selected as the recommended phase 2 dose, with the option to add a future cohort with 25 mg lenalidomide (supplemental Methods). The phase 2 portion of this study started concurrently with the phase 1b cohort 3 still ongoing. Among 17 patients treated in cohorts 2 and 3 (20-mg and 25-mg lenalidomide dose levels), no DLTs were identified and 3 patients discontinued treatment because of a TEAE of worsening DLBCL, altogether suggesting a manageable safety profile for the combination in this population.
The safety profile of the triplet ibrutinib, lenalidomide, and rituximab is consistent with the known individual drug profiles. For ibrutinib, the most common AEs (≥20% of patients) across prior B-cell malignancy studies were neutropenia, thrombocytopenia, diarrhea, anemia, musculoskeletal pain, rash, nausea, bruising, fatigue, hemorrhage, and pyrexia; the most common grade 3 to 4 AE was neutropenia.7 As neutropenia is a known adverse effect of ibrutinib and lenalidomide and has been observed with rituximab,7,9,17 the frequency of neutropenia in this study was not unexpected, and neutropenia is generally manageable with growth factors. In this study, rash was overall less of a concern compared with the triple therapy in FL.21 This may reflect the difference in immunosuppression in the more heavily pretreated population in this study.
In response to the DLTs and AEs seen in cohort 1, and subsequent discussion between the study sponsor and investigators, AE management strategies and revised DLT definitions were implemented via protocol amendments (supplemental Methods). Management strategies included dose delays and/or reductions of study drugs and medical management (corticosteroids and antihistamines for rash; growth factors for neutropenia). The effect of the toxicity management strategies is reflected in the lower incidence of DLTs in cohorts 1+, 2, and 3 vs cohort 1 (Table 3); however, with an increased frequency of dose delays and/or reductions resulting from AEs in cohorts 1+, 2, and 3 (ibrutinib, 78%-100%; lenalidomide, 44%-100%) compared with cohort 1 (ibrutinib, 50%; lenalidomide, 42%). These strategies enabled most patients experiencing these AEs to continue study treatment. The 3 patients who discontinued treatment because of rash or neutropenia experienced DLTs in cohort 1, when the study protocol still demanded patients with a DLT discontinue treatment.
Promising activity was observed for combination ibrutinib and rituximab with doses at least 15 mg lenalidomide in adults with relapsed/refractory DLBCL. Of note, 69% of patients had stage IV disease, 84% had received at least 2 prior treatment regimens, and 27% had primary refractory disease. The presence of only 2 responses in the 20-mg lenalidomide cohort is unexpected, given the responses at the 15-mg and 25-mg lenalidomide doses, and may be a result of large variation from the small number of patients in this cohort. The ongoing phase 2 portion with 20 mg lenalidomide will provide a more robust efficacy assessment at this dose.
In this study (N = 45), ORR with the triplet of ibrutinib, lenalidomide, and rituximab was 38% and 44% in the overall population and response-evaluable patients, respectively. For patients with non-GCB DLBCL, the ORR was 48% and 65%, respectively. In a phase 2 trial of lenalidomide plus rituximab in patients with relapsed/refractory B-cell lymphomas (median age, 66 years; median follow up, 9.8 months), the 32 patients with relapsed/refractory DLBCL had an ORR of 28%.25 In contrast, an observational study in elderly patients (median age, 79 years) treated with lenalidomide plus rituximab reported that 50% of patients achieved either CR or PR (n = 3 each), with a median PFS of 6 months (median follow-up, 12 months); however, this was a small trial of 12 patients, 5 of whom were older than 80 years.26
ORR was noticeably higher among response-evaluable patients with non-GCB DLBCL (65%, including 41% with CR) than in those with GCB DLBCL (29%, including 18% with CR), although this study was not powered for comparisons between subpopulations. Similarly, median DOR was longer in patients with non-GCB vs GCB DLBCL (15.9 vs 8.8 months, respectively). For ABC (n = 7), the response rate was 43%, and for GCB (n = 17), the response rate was 29%. These findings are noteworthy because responses are traditionally higher with salvage chemotherapy in patients with GCB vs non-GCB DLBCL,2,27,28 as the most common subtype (2/3 cases) in elderly patients.29 ORR (46%) and DOR (12.3 months) among response-evaluable patients with transformed DLBCL were also promising. Ibrutinib and lenalidomide interrupt BCR and MYD88 signaling at different points in the signaling pathways. Dual inhibition may thus increase tumor cell apoptosis, particularly in the more aggressive, actively dividing ABC or non-GCB DLBCL subtypes; however, responses were also seen in patients with transformed DLBCL, who typically have GCB subtype. Fifteen patients had transformed DLBCL, and all previously had FL, except 2 with prior chronic lymphocytic leukemia or marginal zone lymphoma. Two patients with transformed DLBCL from FL had non-GCB subtype (both discontinued early because of DLTs).
Among individuals with relapsed/refractory DLBCL who do not undergo transplantation, median OS is approximately 5 months with standard treatment.4 In this study, median DOR was 15.9 months, and for the overall study population, median PFS was 5.5 months and OS was 9.5 months, with 6 patients still receiving study treatment (range, 19.8-45.0 months).
In conclusion, the triplet of ibrutinib, lenalidomide, and rituximab demonstrated promising activity and a manageable safety profile in patients with relapsed/refractory DLBCL, particularly non-GCB DLBCL, and warrants further evaluation in this patient population with a significant unmet medical need. The ongoing phase 2 portion of this study further evaluates the efficacy and safety of this combination regimen in transplant-ineligible patients with relapsed/refractory non-GCB and ABC DLBCL, including a cohort with 25 mg lenalidomide.
Requests for access to individual participant data from clinical studies conducted by Pharmacyclics LLC, an AbbVie Company, can be submitted through Yale Open Data Access Project site at http://yoda.yale.edu.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank all the patients who participated in the PCYC-1123-CA phase 1b study and their families, as well as the study subinvestigators, research nurses, and coordinators at each study site. Anh Tran provided support with protocol development, study start-up, patient enrollment, and study management and oversight. Medical writing and editorial assistance were provided by Kimberly Brooks and were supported by funding from Pharmacyclics LLC, an AbbVie Company.
This study was sponsored by Pharmacyclics LLC, an AbbVie Company.
Authorship
Contribution: A.G., D.M.B., and J.K.N. designed the study; A.G., R.R., N.G., J.M., D.S.M., N.H.D., M.K., M.D., E.K., and J.R. contributed to the accrual and treatment of patients and data acquisition; J.P., D.M.B., and J.K.N. analyzed the data; and all authors contributed to interpretation of the data, critically reviewed and revised the manuscript, approved the final manuscript for publication, and vouch for data accuracy and completeness.
Conflict-of-interest disclosure: A.G. received research funding through institutional clinical studies and received research funding, consulting fees, speaker’s fees, and other honoraria from Acerta, Genentech, Gilead, Janssen, Kite Pharma, Takeda, and Pharmacyclics LLC, an AbbVie Company, and has stock ownership and a leadership role in Clinical Outcome Tracking Analysis. R.R. received consulting fees from Pharmacyclics LLC, an AbbVie Company, and received research funding from Janssen and Pharmacyclics LLC, an AbbVie Company. N.G. received research funding from Celgene, Genentech, Janssen, SGN, TG Therapeutics, and Pharmacyclics LLC, an AbbVie Company; consulting fees from Celgene, Gilead, Janssen, SGN, TG Therapeutics, and Pharmacyclics LLC, an AbbVie Company; speaker’s fees from AstraZeneca, BMS, Gilead, Janssen, SGN, and Pharmacyclics LLC, an AbbVie Company; and honoraria from AstraZeneca, BMS, Gilead, Janssen, SGN, TG Therapeutics, and Pharmacyclics LLC, an AbbVie Company. J.M. received consulting fees from Gilead, Celgene, Kyowa, Seattle Genetics, Alexion, Bayer, Bristol-Myers Squibb, Janssen, Juno, Kite Pharma, Pfizer, and Pharmacyclics LLC, an AbbVie Company; honoraria from Kyowa and Seattle Genetics; and speaker’s fees from AstraZeneca, Bayer, Gilead, Janssen, Kite Pharma, and Pharmacyclics LLC, an AbbVie Company. D.S.M. has stock ownership in Biogen, Eli Lilly, Gilead, Johnson & Johnson, Merck, Novo Nordisk, Pfizer, Vertex, and Zoetis. N.H.D. received research funding from Eisai and Pharmacyclics LLC, an AbbVie Company. M.K. received research funding from Genentech, Merck, and Pharmacyclics LLC, an AbbVie Company, and consulting fees from Kite Pharma and Incyte. E.K. is employed by Comprehensive Cancer Centers of Nevada. J.P. and J.K.N. are employed by Pharmacyclics LLC, an AbbVie Company; J.P. additionally has stock ownership in AbbVie. D.M.B. has employment and leadership roles with, research funding from, stock ownership in, and travel/accommodation expenses from AbbVie and Pfizer, and has patents/royalties with AbbVie. J.R. received research funding from Celgene, Seattle Genetics, and Pharmacyclics LLC, an AbbVie Company; and received honoraria and consulting fees from AstraZeneca, Celgene, Janssen, and Seattle Genetics. M.D. declares no competing financial interests.
Correspondence: Andre Goy, John Theurer Cancer Center at Hackensack University Medical Center, 92 2nd St, Hackensack, NJ 07601; e-mail: goy.andre@hackensackmeridian.org.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal