

Key Points
Mice with Adamts13−/− or a heterozygous cfh mutation (ie, chfW1206R) do not develop spontaneous TMA.
However, mice carrying Adamts13−/−cfhW1206R or cfhR1206R develop TMA with a significantly increased mortality rate.

Abstract
Severe deficiency of plasma ADAMTS13 activity is the primary cause of thrombotic thrombocytopenic purpura (TTP) whereas overwhelming activation of complement via an alternative pathway results in atypical hemolytic uremic syndrome (aHUS), the prototypes of thrombotic microangiopathy (TMA). However, clinical and pathogenic distinctions between TTP and aHUS are often quite challenging. Clinical reports have suggested that complement activation may play a role in the development of TTP, which is caused by severe deficiency of plasma ADAMTS13 activity. However, the experimental evidence to support this hypothesis is still lacking. Here, we show that mice with either Adamts13−/− or a heterozygous mutation of complement factor H (cfh) at amino acid residue of 1206 (ie, cfhW/R) alone remain asymptomatic despite the presence of occasional microvascular thrombi in various organ tissues. However, mice carrying both Adamts13−/− and cfhW/R exhibit thrombocytopenia, low haptoglobin, increased fragmentation of erythrocytes in peripheral blood smear, increased plasma levels of lactate dehydrogenase activity, blood urea nitrogen, and creatinine, as well as an increased mortality rate, consistent with the development of TMA. Moreover, mice with a homozygous mutation of cfh (ie, cfhR/R) with or without Adamts13−/− developed severe TMA. The mortality rate in mice with Adamts13−/−cfhR/R was significantly higher than that in mice with cfhR/R alone. Histological and immunohistochemical analyses demonstrated the presence of disseminated platelet-rich thrombi in terminal arterioles and capillaries of major organ tissues in these mice that were either euthanized or died. Together, our results support a synergistic effect of severe ADAMTS13 deficiency and complement activation in pathogenesis of TMA in mice.
Introduction
Thrombotic microangiopathy (TMA) is a heterogeneous group of thrombotic disorders characterized by severe thrombocytopenia and microangiopathic hemolytic anemia with various degrees of terminal organ injury.1,-3 TMA may develop following infection with Shiga toxin–producing Escherichia coli (STEC), resulting in hemolytic uremic syndrome (HUS), known as STEC-HUS.4,5 TMA may also be caused by the lack of a plasma metalloprotease ADAMTS13 activity, rendering it incapable of cleaving ultra-large von Willebrand factor (ULVWF) released from activated or injured endothelial cells. This type of TMA is known as thrombotic thrombocytopenic purpura (TTP).6,,-9 Furthermore, TMA may be caused by uncontrolled activation of the complement system, most commonly via its alternative complement pathway. This type of TMA is referred to as atypical HUS (aHUS).10,11
STEC-HUS, commonly seen in children under 5 years of age, accounts for 90% of all cases of HUS, resulting primarily from E coli O157 infection.5,12,13 Upon infection with STEC, Shiga toxin A and B are produced in the gut and then enter into the circulation where they trigger acute inflammation, release of ULVWF, and loss of thrombomodulin on the endothelial surface, resulting in a prothrombotic state.14,-16 aHUS, seen in both children and adults, is primarily caused by excessive activation of the complement system via its alternative pathway. This may take place when the function of complement-regulatory proteins is compromised, such as in cases with mutations in complement factor H (CFH),17,18 factor I (CFI),17,19 and membrane cofactor protein (MCP),17,20,21 as well as thrombomodulin22 or when a mutation in C323,24 or factor B (CFB)25,26 occurs, which results in resistance to inhibition by the complement-regulatory proteins.
In humans, the disease penetrance for aHUS with a known heterozygous mutation in CFH (or CFI) is only 50%27 ; in mice, a heterozygous mutation of cfh at the amino acid residue of 1206 (ie, cfhW/R), which corresponds to the CFH mutation (W/R) at the amino acid residue of 1183 in patients with aHUS,28 does not result in a symptomatic aHUS as we previously reported.29 This suggests that additional environmental or genetic factors may be necessary to trigger the onset and result in progression of aHUS.
Approximately 95% of TTP cases are caused by acquired autoantibodies against ADAMTS13 protease8,30 ; only less than 5% of TTP cases are caused by inherited mutations in ADAMTS13.7,31,32 Similar to aHUS, some children with hereditary deficiency of ADAMTS13 develop their first acute episode early in life, but others may develop their first episode of TTP in adulthood, usually triggered by infections or pregnancy.31,32 In almost every case of TTP, there are evidences of neutrophil and complement activation, indicated by the marked elevation of plasma levels of human neutrophil peptides (HNPs),33 DNA, myeloperoxidase,34 histone-DNA complexes,33 Bb, C3a, C3b, C5a, and sC5b-9.33,35,,-38 Some patients with refractory TTP caused by severe ADAMTS13 deficiency appeared to respond to treatment with eculizumab,39,-41 a monoclonal antibody against complement C5, which is shown to block the formation of terminal membrane attack complexes. These results allow us to hypothesize that there may be a synergy between ADAMTS13 deficiency and complement activation in pathogenesis of TMA.
To test this hypothesis, we took advantage of 2 existing mouse lines, Adamts13−/− (Motto et al42 ) and cfhW/R (Ueda et al29 ), to generate a new strain of mice with a combined mutation in Adamts13 and cfh. Our results demonstrate that mice with either Adamts13−/− or cfhW/R remain asymptomatic, but mice with cfhW/R in the Adamts13−/− background exhibit thrombocytopenia, elevated lactate dehydrogenase (LDH) and creatinine, and as well, an increased mortality rate, consistent with the development of TMA. Additionally, mice with cfhR/R on top of Adamts13−/− exhibit more severe organ damage and increased mortality than cfhR/R alone. Our results suggest a synergistic effect of severe ADATMS13 deficiency and complement activation in the pathogenesis of TMA. These findings may provide a rationale for a more targeted therapeutic intervention in patients with refractory TMA.
Methods
Animals
All animal experimental protocols were approved by the institutional animal care and use committee at the University of Alabama at Birmingham. All mutant mice were derived from wild-type (wt) C57BL/6J. Adamts13−/− mice (kindly provided by David Ginsburg, University of Michigan, Ann Arbor, MI)42 were bred with mice carrying a heterozygous cfh mutation (cfh; ie, cfhW/R)29 to generate heterozygous mutations in both Adamts13 and cfh (ie, Adamts13+/−cfhW/R) mice. These mice were then bred to generate mice with various combinations of Adamts13 and cfh mutations (ie, Adamts13−/−cfhW/R and Adamts13−/−cfhR/R). These mice were then compared phenotypically with the age- and sex-matched control mice (eg, wt, Adamts13−/−, cfhW/R, and cfhR/R) in the same genetic background.
Genotyping
All phenotypical data were collected prior to the genotyping or blinded with the genotypes. Genotyping for Adamts13 was performed by polymerase chain reaction amplification with 4 pairs of primers according to the strategy described previously.42,43 For cfh genotyping, Sanger sequencing was performed in the amplified polymerase chain reaction product (forward primer, ATTGACCAGCTACAGACAGTATCA; reverse primer, CATGCATGTGCCTTTCTAA-ACA).
CBCs and erythrocyte morphology analysis
Whole blood was collected via retro-orbital sinus into a heparin-coated capillary tube and anticoagulated with 0.32% sodium citrate. Complete blood counts (CBCs) were performed using a Hemavet 950FS analyzer (Drew Scientific, Miami Lakes, FL).43 Additionally, a thin blood smear was prepared for each sample and fixed in methanol, and stained with Wright-Giemsa stain (Sigma-Aldrich, St. Louis, MO).
Assay for plasma haptoglobin
Plasma haptoglobin was determined using a murine-specific haptoglobin enzyme-linked immunosorbent assay (ELISA) (Abcam, Cambridge, MA).44
Assays for plasma blood urea nitrogen, creatinine, and LDH
Plasma urea nitrogen was determined using a urea nitrogen colorimetric detection assay (Invitrogen, Frederick, MD). Plasma creatinine was determined using a creatinine colorimetric assay (Sigma-Aldrich).45 Plasma LDH levels were determined using an LDH activity assay (Sigma-Aldrich).46 All assays were performed on plasma according to the manufacturers’ instructions.
Assay for plasma soluble C5b-9 complexes
Plasma soluble C5-9 complexes (sC5b-9) levels were determined using the High Sensitivity sC5-9 ELISA kit according to the manufacturer’s recommendation (Aviva Systems Biology, San Diego, CA).
Plasma VWF antigen and multimer analysis
Murine plasma von Willebrand factor (VWF) antigen levels were quantified by an in-house ELISA as described previously.29 Plasma VWF multimers were determined by western blotting following electrophoresis on 1% agarose gel and capillary transferring to membrane. The western blot was performed with a rabbit anti-human VWF immunoglobulin G (IgG; Dako, Glostrup, Denmark) as the primary antibody and IRDye800-labeled goat anti-rabbit IgG (LI-COR Biosciences, Lincoln, NE) as the secondary antibody.47
Histology and immunohistochemical studies
Major organ tissues were harvested from mice of various genotypes when they died or when they were euthanized at the age of ∼2 months. The tissues (brain, heart, intestine, kidney, liver, and lung) were fixed in 4% paraformaldehyde in phosphate-buffered saline and were then paraffin-embedded. Thin sections (6 µm) were prepared using a microtome and stained with hematoxylin and eosin (H&E). Additionally, the adjacent sections were used for immunohistochemical staining for VWF, glycoprotein IIb/IIIa (GPIIb/IIIa), and fibrinogen using a rabbit anti-human VWF IgG (Dako), a mouse anti-human GPIIb/IIIa antibody (a gift from Heyu Ni, University of Toronto, Toronto, ON, Canada), and a rabbit anti-human fibrinogen IgG (Dako), respectively, followed by the peroxidase-conjugated secondary antibody and color reaction with 3,3′-diaminobenzidine/H2O2. Digital images were taken using a Carl Zeiss Axioplan light microscope (Göttingen, Germany) at magnification of ×200.
Statistical analysis
All data were expressed as the median in a box-whisker plot (minimum to maximum values) or as the mean ± a standard deviation (SD). The Mann-Whitney U test was used for comparison between 2 groups. A 1-sample Student t test between proportions was performed to determine whether there was a statistically significant difference between the expected percentage and the observed percentage of certain genotype using MedCalc software (Ostend, Belgium). P < .05 was considered to be statistically significant. The death of affected mice was monitored daily for 180 days. A Kaplan-Meier survival plot and statistical analysis were performed using GraphPad Prism 7 (GraphPad, La Jolla, CA).
Results
Distribution of various genotypes in mice
Table 1 shows the expected and the observed distribution of mice with various genotypes at 4 weeks of age after crossing Adamts13−/−cfhW/W with Adamts13−/−cfhW/R or Adamts13−/−cfhW/R with Adamts13−/−cfhW/R mice. As shown, the percentage of mice with Adamts13−/−cfhW/R (37.5%) was significantly lower than the expected percentage of 50% (P = .01) (Table 1, top panel). Additionally, as the percentage of Adamts13−/−cfhW/W mice was significantly higher (P = .01) than the expected percentage of 25% based on the Mendelian distribution, the percentage of mice with the Adamts13−/−cfhW/R (45.2%) and Adamts13−/−cfhR/R (22.3%) genotype was also modestly lower than the expected 50% and 25%, respectively (Table 1, bottom panel). However, these differences were not statistically significant. These results suggest that only a small subset of mice carrying the heterozygous or homozygous mutation in cfh on top of Adamts13−/− may be lethal in utero or expire shortly after birth.
Expected and obtained distribution of mice with various genotypes
| Genotype | N | Expected, % | Observed, % |
|---|---|---|---|
| a13−/−cfhW/W× a13−/−cfhW/R | |||
| a13−/−cfhW/W | 60 | 50 | 62.5* |
| a13−/−cfhW/R | 36 | 50 | 37.5* |
| a13−/−cfhW/R× a13−/−cfhW/R | |||
| a13−/−cfhW/W | 64 | 25 | 32.5 |
| a13−/−cfhW/R | 89 | 50 | 45.2 |
| a13−/−cfhR/R | 44 | 25 | 22.3 |
| Genotype | N | Expected, % | Observed, % |
|---|---|---|---|
| a13−/−cfhW/W× a13−/−cfhW/R | |||
| a13−/−cfhW/W | 60 | 50 | 62.5* |
| a13−/−cfhW/R | 36 | 50 | 37.5* |
| a13−/−cfhW/R× a13−/−cfhW/R | |||
| a13−/−cfhW/W | 64 | 25 | 32.5 |
| a13−/−cfhW/R | 89 | 50 | 45.2 |
| a13−/−cfhR/R | 44 | 25 | 22.3 |
A Student t test was performed for comparing the difference between the percent observed and expected. The null hypothesis value (percentage expected) is based on the Mendelian distribution.
a13−/−, Adamts13 null; cfh, complement factor H; cfhW/W, cfhW/R, and cfhR/R represent wild-type, heterozygous, and homozygous mutations in the gene encoding cfh at the position of 1206.
P < .05.
Platelet counts in mice with various genotypes
A CBC was performed in each of the adult mice who survived to the age of 2 to 3 months. Platelet counts have been shown to be the most sensitive marker for the diagnosis of TMA in mice.43,48 As shown, there were significantly lower median platelet counts in mice with cfhR/R (662 × 109/L) than in those with cfhW/R (893 × 109/L) (P = .0018) and in wt mice (861 × 109/L) (P = .0042) (Figure 1A). Similarly, there were significantly lower median platelet counts in mice with Adamts13−/−cfhW/R (791 × 109/L) or Adamts13−/−cfhR/R (553 × 109/L) than in mice with cfhW/R (893 × 109/L) or Adamts13−/− (821 × 109/L) (Figure 1A). However, there was no statistically significant difference in the median platelet counts between the cfhR/R mice (662 × 109/L) and the Adamts13−/−cfhR/R mice (553 × 109/L) (Figure 1A). Peripheral blood smears with Wright-Giemsa stain showed the presence of fragmented red blood cells (or schistocytes) in Adamts13−/−cfhW/R, cfhR/R, and Adamts13−/−cfhR/R mice (Figure 1B), but not in any wt control mice or in those carrying Adamts13−/− or cfhW/R (not shown). These results demonstrate that mice with either cfhW/R or Adamts13−/− alone remain asymptomatic, but mice with Adamts13−/−cfhW/R , cfhR/R, or Adamts13−/−cfhR/R develop symptomatic and laboratory-confirmed TMA.
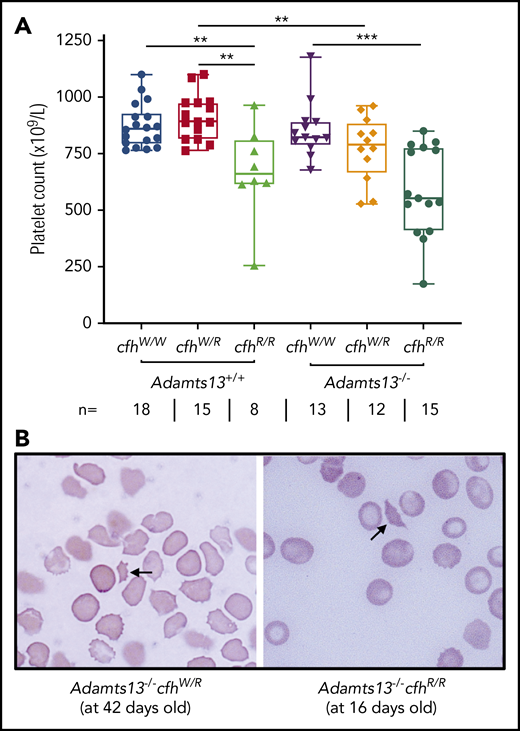
Platelet counts and schistocytes in theperipheral blood smear from mice with various genotypes. (A) Platelet counts in mice with various genotypes are shown as the dots in the box-whisker plots. Each dot represents the platelet count of an individual mouse. The box spans the interquartile range. The top and bottom lines outside of the box represent the maximal and minimal values, respectively. The Mann-Whitney U test was performed to compare the difference of platelet counts in wt or Adamts13−/− with each of the other experimental groups. **P < .01 and ***P < .005, respectively. The number (n =) of mice with each genotype is shown below the graph. (B) Blood smear reveals the red blood cell morphology. The arrow indicates the fragmentated red blood cells (or schistocytes) in the background of other red blood cells from Adamts13−/−cfhW/R at the age of 42 days and Adamts13−/−cfhR/R at the age of 16 days. Wright-Giemsa stain; original magnification ×200.
Platelet counts and schistocytes in theperipheral blood smear from mice with various genotypes. (A) Platelet counts in mice with various genotypes are shown as the dots in the box-whisker plots. Each dot represents the platelet count of an individual mouse. The box spans the interquartile range. The top and bottom lines outside of the box represent the maximal and minimal values, respectively. The Mann-Whitney U test was performed to compare the difference of platelet counts in wt or Adamts13−/− with each of the other experimental groups. **P < .01 and ***P < .005, respectively. The number (n =) of mice with each genotype is shown below the graph. (B) Blood smear reveals the red blood cell morphology. The arrow indicates the fragmentated red blood cells (or schistocytes) in the background of other red blood cells from Adamts13−/−cfhW/R at the age of 42 days and Adamts13−/−cfhR/R at the age of 16 days. Wright-Giemsa stain; original magnification ×200.
Plasma levels of BUN, creatinine, and LDH in mice with various genotypes
To assess the potential organ damage resulting from TMA, we determined plasma levels of blood urea nitrogen (BUN), creatinine, and LDH in mice with various genotypes at the age of ∼2.5 months (or 10 weeks). As shown, the median levels of plasma BUN increased significantly in mice with Adamts13−/−cfhW/R (27.5 mg/dL), cfhR/R (28.2 mg/dL), and Adamts13−/−cfhR/R (28.9 mg/dL) when compared with the levels in wt mice (20.5 mg/dL) or in those carrying Adamts13−/− alone (23.0 mg/dL) (Figure 2A). Interestingly, the median levels of plasma creatinine were only significantly elevated in mice with Adamts13−/−cfhW/R (0.28 mg/dL) and Adamts13−/−cfhR/R (0.31 mg/dL), but not in those with any other genotype (Figure 2B). There was a statistically significant correlation between platelet counts and plasma BUN levels in mice with Adamts13−/−cfhW/R or Adamts13−/−cfhR/R mice (P = .01) (Figure 2F). However, no correlation between platelet counts and plasma creatinine levels was detected in mice with any genotype (not shown). These results suggest that plasma BUN levels may be a more sensitive and reliable marker for assessing the overall renal functions in mice, consistent with results reported previously.29
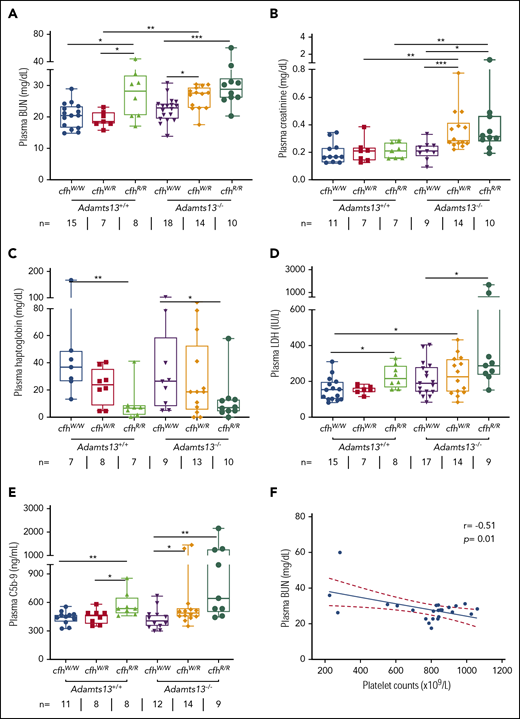
Plasma BUN, creatinine, haptoglobin, and LDH in mice with various genotypes. Box-whisker and dot plots show the median, interquartile, minimum to maximum, and each individual value of plasma BUN (A), creatinine (B), haptoglobin (C), and LDH (D), as well as sC5b-9 (E) in mice at the age of 2 to 3 months with various genotypes. The Mann-Whitney U test was performed to compare the difference in each biomarker between the wt or Adamts13−/− and each of the other experimental groups. (E) A negative correlation was detected between BUN and sC5b-9 in plasma of Adamts13−/−cfhW/R and Adamts13−/−cfhR/R mice. *P < .05, **P < .01, and ***P < .001, respectively. The number of mice (n =) from each genotype tested is shown under each graph. (F) Pearson correlation coefficient (r = −0.51, P = .01) between platelet counts and plasma BUN levels was determined using Prism 7 software.
Plasma BUN, creatinine, haptoglobin, and LDH in mice with various genotypes. Box-whisker and dot plots show the median, interquartile, minimum to maximum, and each individual value of plasma BUN (A), creatinine (B), haptoglobin (C), and LDH (D), as well as sC5b-9 (E) in mice at the age of 2 to 3 months with various genotypes. The Mann-Whitney U test was performed to compare the difference in each biomarker between the wt or Adamts13−/− and each of the other experimental groups. (E) A negative correlation was detected between BUN and sC5b-9 in plasma of Adamts13−/−cfhW/R and Adamts13−/−cfhR/R mice. *P < .05, **P < .01, and ***P < .001, respectively. The number of mice (n =) from each genotype tested is shown under each graph. (F) Pearson correlation coefficient (r = −0.51, P = .01) between platelet counts and plasma BUN levels was determined using Prism 7 software.
To assess the degree of hemolysis, we determined the plasma levels of haptoglobin, which binds free hemoglobin and results in accelerated clearance from circulation during hemolysis.49,-51 As shown, the median plasma haptoglobin levels in mice with cfhR/R (6.9 mg/dL; P = .007) or Adamts13−/−cfhR/R (7.2 mg/dL; P = .03) were significantly lower than those in wt (36.9 mg/dL) or Adamts13−/− (26.5 mg/dL). However, there was no statistically significant difference in the median haptoglobin levels between the cfhR/R mice and the Adamts13−/−cfhR/R mice or between the cfhW/R and Adamts13−/−cfhW/R groups (Figure 2C). These results suggest that complement overactivation may be the primary driving force causing hemolysis.
To assess the degree of overall organ damage, plasma levels of LDH were also determined. High LDH is predictive for more severe disease52 and a higher mortality rate in patients with TTP.53 As shown, there was a significant increase in the median plasma levels of LDH in mice with cfhR/R (216.6 U/L; P = .02), Adamts13−/−cfhW/R (229.2 U/L; P = .04), and Adamts13−/−cfhR/R (283.3 U/L; P = .001) when compared with those in wt mice (153.1 U/L). The highest plasma levels of LDH were detected in mice with Adamts13−/−cfhR/R (Figure 2D). These results indicate that the most severe organ damage occurs in mice with a combined abnormality (ie, severe ADAMTS13 deficiency and a homozygous mutation in cfh).
To assess the status of complement activation in vivo, plasma levels of the soluble terminal complement complex (sC5b-9) were determined in mice with various genotypes. Serum or plasma sC5-9 has been shown to be a reliable marker of complement activation in patients with aHUS54,55 and TTP.36,53,56 The median plasma levels of sC5b-9 in cfhR/R mice (536.6 ng/mL) were significantly higher than those in cfhW/R (461.5 ng/mL; P = .03) or cfhW/W (449.4 ng/mL; P = .005) mice. Moreover, the median plasma levels of sC5b-9 in mice with Adamts13−/−cfhW/R (491.4 ng/mL; P = .04) or Adamts13−/−cfhR/R (644.0 ng/mL; P = .003) were also significantly higher than those in Adamts13−/− mice (406.9 ng/mL). These results suggest that there is more complement activation in mice with a combined abnormality (a mutation in cfh and Adamts13−/−) than in those with a mutation in either cfh or Adamts13 alone.
Plasma VWF antigen and multimers in mice with various genotypes
VWF plays an essential role in pathogenesis of TTP.2,6,57 However, the role of VWF in pathogenesis of aHUS is less well understood.4 Our results showed that plasma levels of VWF antigen (the mean ± SD) were dramatically increased in mice with cfhR/R (183.7% ± 42.4%; P = .0003), Adamts13−/−cfhW/R (132.7% ± 30.4%; P = .02), and Adamts13−/−cfhR/R (143.5% ± 24.1%; P = .0019) compared with those in wt mice (99.5% ± 14.5%) (Figure 3A). No significant difference in plasma VWF antigen levels was detected between Adamts13−/− and wt groups. Interestingly, plasma VWF multimer analysis revealed that the increase of plasma VWF antigen in the cfhR/R mice was primarily attributed by the increase of small to medium sizes of VWF multimers. Few or no ULVWF multimers were detected in mice with cfhW/R or cfhR/R when normal plasma ADAMTS13 activity was present (Figure 3B-D). This may be due to the increase in degradation, proteolysis, or consumption of ULVWF in the ongoing thrombosis. In the absence of ADAMTS13 activity, however, the newly released ULVWF may remain anchored on the endothelial surface. However, there was no significant change in the ratio of high- to low-molecular-weight VWF multimers in mice with Adamts13−/−cfhW/Ror Adamts13−/−cfhR/R compared with that in Adamts13−/− or wt mice (Figure 3E-G). These results indicate that there may be cross talk between ADAMTS13, VWF, and complement activation that regulates endothelial function and thrombus formation.
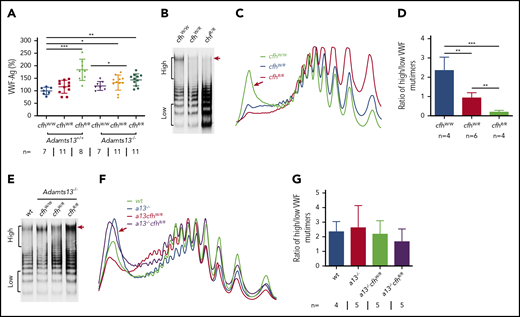
Plasma VWF antigen and multimer distribution in mice with various genotypes. (A) Plasma levels of VWF antigen in mice with various genotypes. Dots and horizontal lines represent the individual, the mean values, and ± SD. The Mann-Whitney U test was performed to compare the difference of VWF antigen between the wt or Adamts13−/− mice with each of the other experimental groups. Representative images of (B,E) VWF multimers, (C,F) densitometric scanning, and (D,G) the ratios of high to low molecular VWF forms, respectively, in mice with various genotypes. The data in panels D and G are the mean ± SD. Mann-Whitney U analysis was performed to determine the statistical significance among various groups. *P < .05, **P < .01, and ***P < .005, respectively.
Plasma VWF antigen and multimer distribution in mice with various genotypes. (A) Plasma levels of VWF antigen in mice with various genotypes. Dots and horizontal lines represent the individual, the mean values, and ± SD. The Mann-Whitney U test was performed to compare the difference of VWF antigen between the wt or Adamts13−/− mice with each of the other experimental groups. Representative images of (B,E) VWF multimers, (C,F) densitometric scanning, and (D,G) the ratios of high to low molecular VWF forms, respectively, in mice with various genotypes. The data in panels D and G are the mean ± SD. Mann-Whitney U analysis was performed to determine the statistical significance among various groups. *P < .05, **P < .01, and ***P < .005, respectively.
Kaplan-Meier survival analysis in mice with various genotypes
Mice carrying Adamts13−/− or cfhW/R alone were viable, fertile, and largely asymptomatic. There was a small increase in the mortality rate in Adamts13−/− mice (4%) compared with wt (0%) or cfhW/R (0%) mice. However, the mortality rate in mice with Adamts13−/−cfhW/R (14.2%) was significantly higher than that in mice with Adamts13−/− or cfhW/R alone. Likewise, the mortality rate in mice with Adamts13−/−cfhR/R (58.3%) was dramatically increased compared with that in mice with Adamts13−/− (4%) or cfhR/R (42.1%) alone (Figure 4). These results suggest that individuals carrying either cfhW/R or cfhR/R on top of severe ADAMTS13 deficiency may have significantly higher mortality rates than those carrying a single gene mutation. This is at least the case in mice.
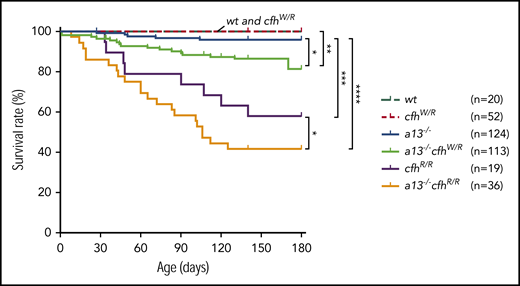
Kaplan-Meier survival analysis of mice with various genotypes. The mortality rate (percentage) was determined for 6 months in mice with various genotypes. The log-rank (Mantel-Cox) test was used to compare the difference in the mortality rate between the control group (wt or cfhW/R) and each of other genotypes. *P < .05, **P < .01, ***P < .005, and ****P < .0001, respectively. a13−/−, Adamts13−/−; cfhW/R and cfhR/R are the heterozygous and homozygous mutations, respectively.
Kaplan-Meier survival analysis of mice with various genotypes. The mortality rate (percentage) was determined for 6 months in mice with various genotypes. The log-rank (Mantel-Cox) test was used to compare the difference in the mortality rate between the control group (wt or cfhW/R) and each of other genotypes. *P < .05, **P < .01, ***P < .005, and ****P < .0001, respectively. a13−/−, Adamts13−/−; cfhW/R and cfhR/R are the heterozygous and homozygous mutations, respectively.
Histology and immunohistochemical analysis of major tissues in mice with various genotypes
To determine the extent of microvascular thromboses and thrombus compositions, we performed H&E histology analysis and immunohistochemistry on tissue sections from brain, heart, lung, liver, and kidney, etc. As shown, the occlusive microvascular thrombi stained with H&E (Figure 5, left column) and antibodies against platelet integrin β3 (Figure 5, middle column) or fibrinogen (Figure 5, right column) were present in all tissues examined except for spleen obtained from mice with Adamts13−/−cfhW/R (Figure 5) but more prominent in the tissues from mice with cfhR/R or Adamts13−/−cfhR/R (not shown). Occasional microvascular thrombi (1-2 thrombi per 10 high-power fields) were also detected in the brain, heart, kidney, and intestine in the asymptomatic mice (n = 3) with Adatms13−/− or cfhW/R alone (not shown). No occlusive thrombus was detected in any organ tissue in the wt mice. All control slides without the primary antibody were stained negative. These results suggest that silent thrombi may occur in the asymptomatic mice carrying Adamts13−/− or cfhW/R, but disseminated occlusive thrombi are only present in mice with Adamts13−/−cfhW/R, cfhR/R, and Adamts13−/−cfhR/R, which is confirmatory for TMA in these mice.
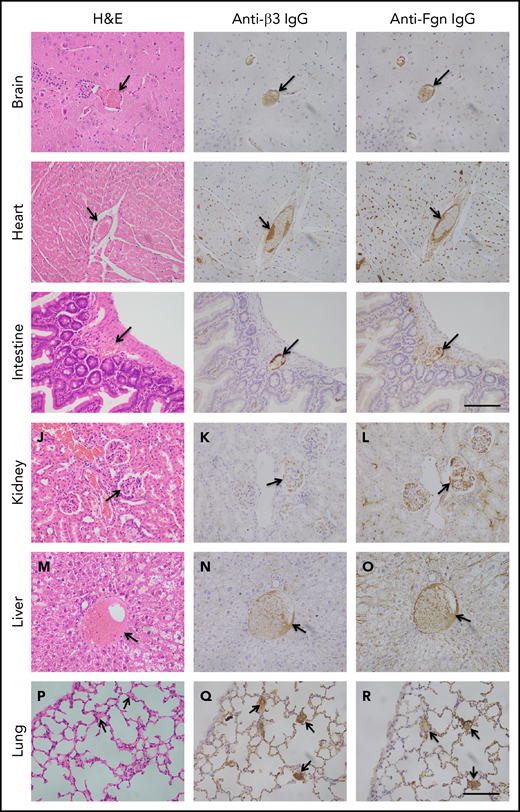
Histology and immunohistochemical analysis of microvascular thrombosis in mouse tissues. Representative images of major organ tissues from Adamts13−/−cfhW/R at the age of 3 months being euthanized, including brain (A-C), heart (D-F), intestine (G-I), kidney (J-L), liver (M-O), and lung (P-R), stained with H&E (left column), anti-β3 IgG (middle column), and anti-fibrinogen (Fgn) IgG (right column) as described in “Methods.” Arrow indicates the presence of occlusive and platelet-rich microvascular thrombus. Scale bar, 100 μm.
Histology and immunohistochemical analysis of microvascular thrombosis in mouse tissues. Representative images of major organ tissues from Adamts13−/−cfhW/R at the age of 3 months being euthanized, including brain (A-C), heart (D-F), intestine (G-I), kidney (J-L), liver (M-O), and lung (P-R), stained with H&E (left column), anti-β3 IgG (middle column), and anti-fibrinogen (Fgn) IgG (right column) as described in “Methods.” Arrow indicates the presence of occlusive and platelet-rich microvascular thrombus. Scale bar, 100 μm.
Discussion
The present study demonstrates a potential synergistic role of severe ADAMTS13 deficiency and overactivation of complement system in a murine model of TMA. As we and others have previously reported, mice with only cfhW/R (Ueda et al29 ) or Adamts13−/− (Motto et al42 and Jin et al48 ) are asymptomatic with no thrombocytopenia or elevated LDH except for occasional thrombi on tissue examination. However, mice with cfhR/R or Adamts13−/−cfhW/R develop symtomatic TMA, indicated by low platelet counts, low haptoglobin, increased fragmentation of red blood cells, and increased LDH, BUN, and creatinine. Disseminated microvascular thrombi are present in nearly all organ tissues in these mice, further confirming the diagnosis. Moreover, mice with Adamts13−/−cfhW/R or Adamts13−/−cfhR/R have an increased mortality rate when compared with those carrying cfhW/R or cfhR/R or Adamts13−/− alone. Together, these results demonstrate a potential synergistic, albeit detrimental, effect of severe ADAMTS13 deficiency and heightened complement activation in the onset, progression, and outcome of TMA in mice.
TTP/HUS was historically considered to be 1 complicated syndrome.58,59 With the discovery of ADAMTS13,7,60,-62 TTP is now recognized as a distinct entity, primarily resulting from autoantibodies against ADAMTS13.8,49,53,63,64 An IgG-type autoantibody binds ADAMTS13 and inhibits its activity in >95% of TTP cases.8,30,49,65,,-68 Therapeutic plasma exchange (TPE) plus corticosteroids and rituximab has been the mainstay of treatment of such an immune-mediated TTP (iTTP).69,70 TPE is thought to remove autoantibodies while replenishing the missing or inhibited ADAMTS13 enzyme.8,71 The clinical course in each patient is quite different with some responding to therapy rapidly and completely while others respond to the therapy slowly and unevenly or not at all.72,-74 However, the mortality rate in patients with iTTP remains as high as 10% to 20%.33,75 In those who survive the acute episode, ∼40% of patients may experience an exacerbation and/or relapses despite all proper therapeutics given.33,75,76 With the introduction of caplacizumab, an anti-VWF A1 nanobody, the exacerbation rate has significantly reduced during TPE and after the discontinuation of TPE.76,77 In any case, the heterogeneity in disease severity and responses to therapies suggest the presence of other disease modifiers in the pathogenesis of TTP.
Noris et al reported the presence of a heterozygous mutation in CFH (S890I) in a patient with congenital (cTTP) who has renal failure.78 Chapin et al also reported the presence of heterozygous mutations in CFH in 4 cases with ticlopidine-induced TTP in whom there was severe deficiency of plasma ADAMTS13 resulting from an autoantibody against ADAMTS13.79 More recently, studies have demonstrated that elevated serum or plasma markers of complement activation via an alternative pathway are prevalent in patients with both cTTP and iTTP.35,,-38,56,80,81 Conversely, some pediatric patients with aHUS were found to have a partial82 or even severe deficiency of plasma ADAMTS13 activity,83 although these patients with severe plasma ADAMTS13 deficiency in the absence of anti-ADAMTS13 IgG would have been diagnosed as cTTP rather than aHUS today.70,84 Together, these findings suggest that complement activation and partial or severe deficiency of plasma ADAMTS13 activity could coexist in patients with a clinical diagnosis of aHUS or TTP.
The associations between ADAMTS13, VWF, and complement components or regulators have been studied in vitro. Turner and Moake demonstrate that C3, CFB, CFD, C5, CFH, and CFI are able to attach to the newly released ULVWF strings on the endothelial surface in the amount required for assembly of the alternative pathway components into active complexes.85 Additionally, small VWF multimers are shown to act as a cofactor mediating the CFI-dependent degradation of C3b, thereby inhibiting complement activation, whereas ULVWF multimers do not have such an inhibitory activity toward complement activation, thus, they may promote complement activation.86 When ADAMTS13 is present, the newly released endothelial ULVWF multimers are rapidly and efficiently removed from the cell surface. This will not only reduce platelet adhesion and aggregation but also eliminate the complement component assembly and activation on the injured or activated endothelium. When ADAMTS13 is absent, endothelial ULVWF strings may serve as a template for assembling complement components, which leads to overactivation of complement in situ, resulting in endothelium damage and thrombus formation.
The interaction between CFH and VWF remains elusive. One report demonstrates that CFH may function as a disulfide reductase, which reduces the size of VWF multimers under shear.87 Other reports show that CFH may bind VWF and inhibit88 or enhance its proteolytic cleavage by ADAMTS13.89 Our own unpublished data are consistent with the latter under shear (W.C. and X.L.Z, unpublished results, 14 July 2019). Plasma ADAMTS13 activity in mice with cfhR/R increases by 50% as we previously reported.29 This may at least partially explain the reduction of VWF multimer size in mice with cfhR/R. However, a marked increase of plasma VWF antigen in these mice suggests complement-mediated activation and damage to vascular endothelium. The degree of endothelial injury may be more severe in mice with Adamts13−/−cfhR/R than with cfhR/R based on the increased levels of plasma sC5b-9 and the increased mortality rate in the double mutant mice, despite lower plasma VWF antigen concentrations in these mice. Without a functional ADAMTS13, the released ULVWF in Adamts13−/−cfhR/R mice may remain anchored on the endothelial surface, which promotes activation of complement components and thrombus formation. We conclude that severe deficiency of plasma ADAMTS13 activity and heightened activation of the complement system may play a synergistic, albeit detrimental, role in the onset, progression, and outcomes of TMA in mice.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank David Ginsburg for providing us with Adamts13−/− mice and Dezhi (Annie) Wang at the Core Research Laboratory, Department of Pathology, University of Alabama at Birmingham (Birmingham, AL) for technical support in performing histology and immunohistochemistry. The authors appreciate the gift of mouse anti-human GPIIb/IIIa antibodies from Heyu Ni at the University of Toronto (Toronto, ON, Canada). The authors thank Nicole K. Kocher at the Department of Pathology, University of Alabama at Birmingham for proofreading the manuscript.
This work was partially supported by grants from the National Institutes of Health, National Heart, Lung, and Blood Institute (HL126724) (X.L.Z.), the Answering TTP Foundation (X.L.Z.), and the postdoctoral fellowship grant (18POST33960098) from the American Heart Association (L.Z.).
Authorship
Contribution: L.Z. and X.L.Z. designed research, performed experiments, analyzed the results, and wrote the manuscript; D.Z, and W.C. performed some experiments and analyzed the data; W.-C.S. provided critical reagents; and all authors reviewed and approved the final version of the manuscript.
Conflict-of-interest disclosure: X.L.Z. is a speaker for Alexion, is a consultant/speaker for Sanofi, is the founder of Clotsolution, and received research support from Takeda for a clinical trial using recombinant ADAMTS13. The remaining authors declare no competing financial interests.
Correspondence: X. Long Zheng, Division of Laboratory Medicine, Department of Pathology, The University of Alabama at Birmingham, 619 19th St S, Birmingham, AL 35243; e-mail: xzheng@uabmc.edu.
